6.3 Hands (Bone and Joint Diseases)
6.3.1 Case 116 (Fig. 6.35)
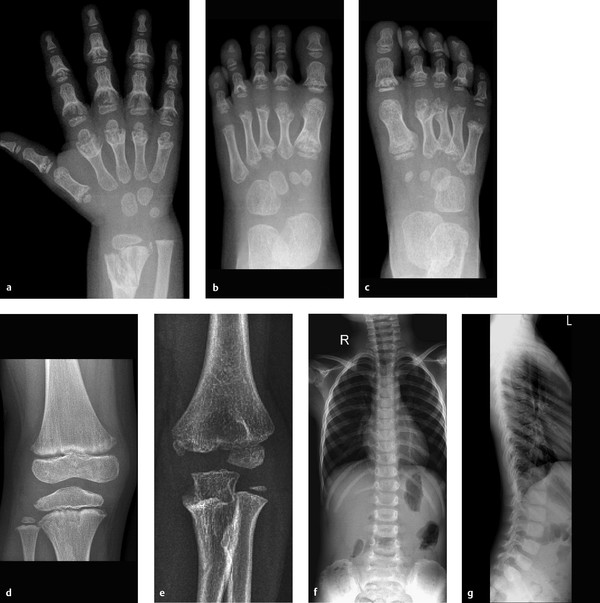
Case description
Referring physician: hand surgeon.
Prior history and clinical question: A 5-year-old child presented with unusual radiologic changes in the metaphyses of the hand skeleton. The child manifested growth retardation and slight bowing of the arms.
Radiologic Findings
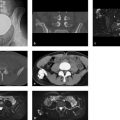
Radiographs of the hands and feet (Fig. 6.35 a–c) show deep cupping of the metaphyses with normally shaped epiphyses. Irregular cartilaginous calcifications are visible within the depressions. The shafts of the small tubular bones appear shortened. The metaphyseal contours about the knee and elbow joints (Fig. 6.35 d, e) appear somewhat irregular but do not show as much cupping as in the hands and feet. The spine appears normal (Fig. 6.35 f, g). Overall, the bones appear slightly more radiolucent than normal.
Location
The pathologic changes described above are confined entirely to the metaphyses, most notably in the hands and feet.
Pathoanatomic Background of the Findings
The changes, limited to the metaphyses, are actually consistent only with skeletal dysplasia based on a chondrocyte defect in the growth plates, resulting in underdeveloped metaphyses.
Assignment to a Possible Basic Entity
Normal variant or malformation?
Based on the considerations above, we are compelled to classify the changes as a developmental abnormality or dysplasia.
Synopsis and Discussion
Once the changes have been classified as a disorder of skeletal development with maldeveloped metaphyses, it is an easy matter to find the disease in a catalog of bone dysplasias. Spranger et al37 described the condition as the “Jansen type of metaphyseal dysplasia.” It is based on mutations of the PTHR1 gene, which is located on chromosome 3p22-p22.1. The mutations lead to the constitutive, ligand-independent activation of the G-protein-coupled receptor for the parathyroid hormone-related protein in pre-hypertrophic chondrocytes of the growth plate. This in turn delays the differentiation of the chondrocytes, leading to a disturbance of bone formation. The principal radiologic changes in children are as follows:
Variable degree of osteopenia (as in our case)
Rickets-like metaphyseal cupping, which may be extreme in early school-age children (as in our case)
Cortical erosions and increased subperiosteal bone (absent in our case)
Calvarial changes (density and structure; unknown in our patient because skull radiographs were not obtained)
Even severe rickets would not cause such deep defects in the metaphyses of the hands and feet.
Final Diagnosis
Jansen type of metaphyseal dysplasia.
Comments
Using an established radiologic hallmark and consulting reference atlases can pave the way to a correct diagnosis.
6.3.2 Case 117 (Fig. 6.36)

Case description
Prior history and clinical question: This case was diagnosed and furnished to use by Dr. C. T. Muth of Cottbus, Germany. It involves a grotesque but painless hypertrophy of the third finger of the left hand in a 73-year-old man, allowing us to dispense with our usual descriptions and deductions. The radiograph of the left hand was obtained after an injury (Fig. 6.36).
Traditionally, the pathogenesis of macrodystrophia lipomatosa is related to the hypertrophy of all mesodermal tissues, but with a dominant fat component, caused by the lipomatosis of a nerve (fibrolipomatous hamartoma). The affected nerve undergoes a “sausagelike” enlargement due to the abnormal proliferation of fibrofatty tissue.46 , 47 The median nerve and its branches or the plantar nerves are most commonly affected. The lipomatosis of the nerve apparently leads to a localized macrodactyly characterized by the hypertrophy of one or more bones and subcutaneous fat in the hand or foot. This hypertrophy is also known as localized gigantism. In very care cases, even large nerves such as the sciatic nerve may be involved.
Based on more recent observations, the dominant fibrolipomatous tissue is found in association with arteriovenous fistulas or circumscribed, hypertrophic vascular structures (arterial, venous, lymphatic). Since macrodystrophia lipomatosa is also thought to be associated with neurofibromatosis type I, it is reasonable to ask whether it may be a subtype of the phacomatoses. One member of this group is proteus syndrome, a hamartoneoplastic syndrome involving all three germ layers. This extremely rare disease is characterized by the following symptoms:
Localized gigantism of the hands and/or feet
Nevi
Hemihypertrophy due to overgrowth of a long tubular bone
Subcutaneous tumors (lipomas, lymphangiomas, hemangiomas)
Macrocephaly
Cranial hyperostosis
Pulmonary and renal anomalies
The disease apparently results from a (somatic) mosaicism. We have seen two cases in which the dominant features were gigantic fat hypertrophy and vascular anomalies (venous malformations, arteriovenous malformations) with hemihypertrophy. One of the cases is published in Freyschmidt (2008).13 Based on current understanding, proteus syndrome is classified among the PTEN hamartoma tumor syndromes (PTEN = phosphate and tensin homolog, deleted on chromosome 10).48
Reportedly, ultrasound and MRI can unequivocally demonstrate the changes of macrodystrophia lipomatosa.46 , 47 Ultrasound shows a diffusely enlarged nerve with alternating hyperechoic (fatty) and hypoechoic bands (nerve bundles) that have a cablelike appearance. MRI shows a similar pattern of longitudinal cylindrical areas of low to intermediate signal intensity (nerve bundles) surrounded by fatty tissue in a diffusely thickened nerve. Patients with macrodactyly also have increased fatty tissue content in the fingers. A proportionate degree of bony hypertrophy is present, as in our case. When cylindrical or serpiginous structures are seen on MRI or CT, we would also consider vascular malformations and try to visualize them with contrast medium. In our case the faint serpiginous structures visible in the soft tissues of the third finger may well represent abnormal vessels.
Final Diagnosis
Macrodystrophia lipomatosa with gigantism of the left third finger.
Comments
Localized gigantism of one finger or toe or multiple fingers or toes should raise suspicion of a hamartoneoplastic syndrome.
6.3.3 Case 118 (Fig. 6.37)

Case description
Referring physician: hand surgeon.
Prior history and clinical question: A 41-year-old woman presented with recurrent pain in her left hand, predominantly on the extensor side. The symptoms were interpreted as tenosynovitis caused by accessory ossicles or heterotopic ossifications. Based on the MRI findings, the surgeon asked if a different diagnosis should be considered.
Radiologic Findings
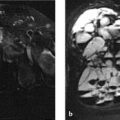
Three well-differentiated bony elements are visible on the dorsal side of the distal row of carpal bones, and a fourth is located deep in the carpal tunnel (Fig. 6.37 a–d; arrows in c, d). These small bony elements are surrounded by thick soft-tissue structures that are isointense to articular cartilage on the T1-image (f) and on the water-sensitive MR sequences (Fig. 6.37 e, g). The second through fourth extensor tendons are significantly displaced.
Location
The epicenter of the osseous and soft-tissue changes appears to be located in the scaphoid.
Pathoanatomic Background of the Findings
It is likely that the “supernumerary” bony elements have something to do with the surrounding thick soft-tissue structures. They may have originated from them. This is also suggested by their differentiated structure with cancellous bone, medullary cavities, and surrounding cortex, resembling the head of an osteochondroma.
Assignment to a Possible Basic Entity
Normal variant or malformation?
The presence of thick, surrounding cartilaginous structures in itself suggests that the supernumerary bony elements are not accessory bones. The same reasoning would rule out heterotopic ossifications. If we follow the line of reasoning under Pathoanatomic Background of the Findings, they may actually represent osteochondromas, although they cannot have originated in the usual way from heterotopic physeal cartilage but from an epiphysis (dysplasia epiphysealis). It is helpful to note that an “irregular” bone such as the scaphoid does not have a classic epiphysis like that found in tubular bones. Instead, the bone itself constitutes an epiphysis consisting of an ossification center surrounded by an acrophysis and covered by articular cartilage. Acrophysis (synonym: spherical growth plate) is a term applied to all other normal growth plates that do not conform to the classic anatomy of an epiphyseal plate. An acrophysis is composed of cartilage cells that are arranged in different zones, as in an ordinary epiphyseal plate:
Zone of resting cartilage
Zone of proliferation
Zone of cartilage column formation
Zone of hypertrophy
Zone of provisional calcification
Trauma?
No trauma history.
Tumor?
No. A cartilage-forming tumor or tumorlike lesion (e.g., synovial chondromatosis, extraskeletal chondrosarcoma) has a more inhomogeneous cartilaginous ossification pattern. In our case we are dealing with very mature bony elements that appear to have been present for a long time.
Synopsis and Discussion
All of the above considerations lead us to epiphyseal osteochondromas, known also as dysplasia epiphysealis or Trevor disease. It may be considered a variant of osteochondroma. This very rare disease more commonly affects the lower limb than the upper limb and is typically unilateral. Cartilage proliferation is usually confined to the medial or lateral side of the affected limb (hemimelic)—in our case the radial side of the carpus. There is also a localized (monostotic) form, a classic form in which more than one bone segment is affected, and a generalized form involving the whole limb. The localized form most commonly affects the bones of the hindfoot or distal tibia, while the classic hemimelic form affects more than one, but not all, epiphyses within a lower limb, most commonly about the knee and ankle joints.
Areas of cartilage proliferation consist of a lobulated mass with a cartilage cap, which develops from the ossified epiphyseal center. Because the scaphoid bone often consists of two growth centers or possibly more, multiple osteochondromas may develop. Conventional radiographs show a well-differentiated bony element that is either located some distance from the associated bone or is adherent to it. The key feature is the surrounding cartilage mantle, which is best demonstrated by MRI. Enchondral ossification may give rise to very dense bone (see Fig. 6.38). If the tendon sheaths are displaced, as in our case, tenosynovitis may develop causing the patient to become symptomatic.
Fig. 6.38 illustrates another case of Trevor disease, also originating from the scaphoid. In this case, a 5-year-old girl, the osteochondroma has developed in a distal and dorsal direction and is attached to the original scaphoid. Portions of the lesion show extremely dense ossification.

Final Diagnosis
Dysplasia epiphysealis hemimelica (Trevor disease) originating from the scaphoid bone.
Comments
Large, differentiated supernumerary, bony elements surrounded by a cartilage mass suggest epiphyseal osteochondroma formation in Trevor disease.
6.3.4 Case 119 (Fig. 6.39)

Case description
Referring physician: surgeon.
Prior history and clinical question: A 32-year-old woman complained of recurrent pain in the bones of her hands and feet. The patient claimed to have experienced increasing forgetfulness and occasional loss of orientation in the past 2 years.
Radiologic Findings
On the plain radiographs, loss of trabecular structure is noted in both carpi, in the metacarpal heads, and in the bases of the proximal phalanges, creating a “cystic” appearance (Fig. 6.39 a, b). The metatarsal heads exhibit similar changes (Fig. 6.39 c). Lateral radiographs of the ankle joints (Fig. 6.39 d, e) show an extreme loss of trabecular structure in all imaged bones, with areas of severe cortical thinning. The CT scans in Fig. 6.39 f, g show fatty tissue in the lucent zones. Attenuation measurements in scans with a soft-tissue window showed attenuation values of –20 to –70 HU. On the T1w MR image in Fig. 6.39 h, the areas showing loss of trabeculation are isointense to fat. Sectional images of the hands yielded similar findings.
Location
The abnormal lucencies in the imaged bone segments are confined to the intraosseous space.
Pathoanatomic Background of the Findings
The “cystic” changes noted in the cancellous bone can be explained by fat replacement. Changes of this kind are found only in polycystic lipomembranous osteodysplasia. This term encompasses the cardinal symptoms described by the patient:
(Pseudo)cystic changes
Fatty tissue
Multifocal bone dysplasia
Assignment to a Possible Basic Entity
Normal variant or malformation?
Yes, in the sense of a developmental disorder. As there is no realistic differential diagnosis for the findings acquired with various modalities (multifocal fat replacement), there is no need to consider other possible categories.
Synopsis and Discussion
The present case of polycystic lipomembranous osteodysplasia could be diagnosed noninvasively by imaging studies alone. CT and MRI were instrumental in proving the fatty nature of the “polycystic” changes.
Polycystic lipomembranous osteodysplasia is generally associated with sclerosing leukoencephalopathy. This combination is called Nasu–Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy) after the authors who first described it. Defined in very general terms, the disease is a “congenital disorder of fatty tissue marked by changes in the bone and central nervous system.” As described above, the pathoanatomic background of the bone changes is a multifocal replacement of normal cancellous bone by fatty tissue. Interspersed among the otherwise normal fat lobules are membranes composed of mucopolysaccharides and lipoproteins that are PAS-positive (PAS = periodic acid-Schiff reaction). Typically, the changes develop symmetrically in the limb bones, especially in the hands and feet but later in the epimetaphyses of the long tubular bones. The sclerosing leukoencephalopathy is marked by a general atrophy and sclerosis of the white matter, particularly in the frontal lobes. Microscopy reveals general diffuse astrogliosis and demyelination. The disease is hereditary and is ascribed to a mutation of the DAP-12 gene.49 A possible link between the osseous and cerebral changes is beyond our present scope, but further details may be found in Freyschmidt (2008).13
Based on our own experience with nine cases, and in agreement with the approximately 200 cases published in the literature,50 symptoms begin at approximately 20 years of age with pain following minor trauma or a greater stress to the affected bones. Pathologic fractures develop by about 30 years of age, with associated onset of neuropsychiatric symptoms that resemble Alzheimer disease. This eventually progresses to frank dementia, and epileptic seizures occur. Most patients die at approximately 50 years of age.
Our patient and her two brothers with the same disease, whom we were able to follow for many years, also developed increasing neuropsychiatric symptoms. Brain MRI in our patient (Fig. 6.39 k, l) showed significant white matter atrophy with ventricular dilatation and diffuse foci of demyelination, which accounted for the clinical symptoms. The skeletal changes progressed at a relatively rapid rate in all three siblings. One brother, for whom we have complete radiologic documentation, developed grotesque fractures of the long bones (Fig. 6.39 i, j) with eventual pathologic fractures in the pelvis and spine.
Final Diagnosis
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (Nasu–Hakola disease).
Comments
Symmetrical cystlike lucencies in the bones of the hands and feet should be investigated further by sectional imaging to determine whether they contain fat. If so, the patient may have polycystic lipomembranous osteodysplasia.
6.3.5 Case 120 (Fig. 6.40)

Case description
Referring physician: hand surgeon.
Prior history and clinical question: A 24-year-old man complained of occasional pain in the left hand. The ulnar head was slightly prominent dorsally and was too far proximal, apparently because the ulna was too short. The surgeon wanted to know the cause of the osteolytic changes in the third and fourth fingers.
Radiologic Findings
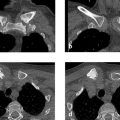
Osteolytic lesions, some expansile (e.g., in the third metacarpal head), are noted in the distal half of the third and fourth metacarpals and associated phalanges (Fig. 6.40 a, b). When the films were viewed with a magnifier, fine calcifications were visible in some of the lesions. The third finger is slightly too long, and the ulnar head on the left side is too far proximal. The distal radial articular surface is sharply sloped toward the ulnar side. The bones of the right hand appear normal.
Location
The pathologic findings are confined to the bones of the left hand.
Pathoanatomic Background of the Findings
When we consider the finding in the distal left radial and ulnar epiphysis, which may be termed a pseudo-Madelung deformity, plus the slightly elongated third finger of the left hand, and the fact that the osteolytic changes are confined to the left hand, we may classify the osteolytic changes in the third and fourth fingers generally as bone dysplasia with foci of cartilage proliferation.
Assignment to a Possible Basic Entity
Normal variant or malformation?
Yes, see Pathoanatomic Background of the Findings and Synopsis and Discussion.
Tumor or tumorlike lesions?
The osteolytic lesions are caused by enchondromas, as indicated by the fine intralesional calcifications. The enchondromas in the ulnar base of the proximal phalanx and in the proximal half of the middle phalanx of the middle finger on the ulnar side are more protuberant than at other sites (enchondroma protuberans). Other possible causes of osteolytic changes in the third and fourth fingers such as granulomas in sarcoidosis, brown tumors of hyperparathyroidism, or gouty tophi do not fit into the above context (unilateral occurrence, signs of dysplasia).
Synopsis and Discussion
The dysplastic changes in the bones of the left hand combined with enchondromas are consistent with the Ollier type of enchondromatosis. The WHO classification of bone tumors15 defines Ollier disease as:
“A developmental disorder caused by a failure of normal enchondral ossification. Furthermore, there is production of cartilaginous masses (enchondromas) in the metaphyses and adjacent regions of the shafts and flat bones, with varying degrees of bone deformity. There is predominant unilateral involvement. The multiple enchondromas appear in childhood and there is widespread skeletal involvement.”
Enchondromatoses do not constitute a uniform group of diseases due to their varied clinical, genetic, and radiologic features. Six different types are distinguished, of which only the Ollier type (unilateral involvement predominantly of the lower limb) and Maffucci syndrome (combines the features of Ollier disease with soft-tissue hemangiomas) are mentioned here. The diseases are generally manifested in early childhood by external deformities and longitudinal growth disturbance. With involvement of the upper limb, the ulna is often shortened relative to the radius, leading to a pseudo-Madelung deformity, as in the present case.
Our own experience with more than 30 cases indicates that enchondromatosis may begin with large, solitary, clinically symptomatic enchondromas and periosteal chondromas, with additional cartilaginous tumors developing much later on the same side of the body. This is significant in that enchondromatoses have an approximately 60% rate of malignant transformation, even at an early age, and therefore patients require life-long clinical and radiologic follow-ups (whole-body MRI every 2 years). Enchondromatosis is not only associated with enchondromas (as the name implies) but also with periosteal chondromas, which are otherwise very rare (see Case 105). Columnar zones of cartilage proliferation appear radiographically as lucent streaks, bands, grooves, or helical lucencies with sclerotic margins in the metadiaphyses (see Case 115). T1w MRI depicts them as linear hypointensities or signal voids which “light up” on T2w images (Fig. 6.41 m, n).
Fig. 6.41 presents the case of an initially 10-year-old boy with a classic form of enchondromatosis, whom we were able to follow for 11 years. The most striking radiographic findings at age 10 were columnar lucencies in the fourth and fifth fingers and in the proximal phalanx of the big toe on the left side (Fig. 6.41 a, b). Eleven years later (Fig. 6.41 c) the skeletal changes in the hand were significantly larger. At that time disseminated osteolytic lesions were found in the left acetabulum (Fig. 6.41 d, j–l), left proximal femur (Fig. 6.41 e, f), distal femoral shaft (Fig. 6.41 g, h), and left distal fibular shaft. The cartilaginous nature of the changes is clearly appreciated on MRI (Fig. 6.41 l–n). This particularly applies to the cartilage columns in the left, lateral distal femoral shaft (Fig. 6.41 m, n). Four small phleboliths (arrows in Fig. 6.41 q) located within a hemangioma (Fig. 6.41 o, p) are seen just above the level of the olecranon and projected over the ulnar epicondyle. These findings were definitive for Maffucci syndrome. Because the enchondromas in the left proximal femur looked particularly aggressive, we obtained several percutaneous CT-guided biopsies at those sites, all of which indicated pure enchondroma with no malignant elements. Since then the young man has presented for 2-year MRI follow-ups, which have shown no significant progression of the changes.


Fig. 6.42 presents another case of enchondromatosis in a 31-year-old man. An unusual feature of this case is the extensive “crossover” skeletal changes involving the left hand and the right lower limb. They appear to be very aggressive in the hand, where the radiograph shows cortical perforations in the affected phalanges. Note that the cartilaginous ossifications in the right femur extend far into the shaft.

Fig. 6.43 shows additional cases of enchondromatosis with grotesque skeletal changes in the hands: a 30-year-old man (Fig. 6.43 a); an incidental finding in a man over the age of 60 (Fig. 6.43 b). It is noteworthy that cartilage masses have perforated into the soft tissues, especially about the middle phalanges. If the patient had reported pain and lesion enlargement at those sites, malignant transformation to chondrosarcoma would have been very likely.

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


