Abdominal masses in children present with variable symptoms, including abdominal pain, abdominal distention, and palpable mass on physical examination. Palpable abdominal masses are a common presenting problem on pediatric outpatient services. Although many pediatric patients ultimately have benign causes for their palpable abdominal masses, such as constipation (in fact, up to 3% of all pediatric outpatient visits are thought to be secondary to constipation), there is a subset of patients who have more significant underlying disease. In addition, malignant masses may present with constitutional symptoms or symptoms related to metastatic disease.
After reasonable measures have been undertaken to exclude benign causes, including collecting a detailed clinical history and performing a physical examination, appropriate imaging should be directed by the physical examination findings, relevant history, and laboratory test abnormalities. Ultrasound (US), with its lack of ionizing radiation, ease of acquisition, and user-friendly environment, is the first-line modality for imaging of most abdominal masses. If the US demonstrates an abdominal mass, additional cross-sectional imaging should be performed. Magnetic resonance imaging (MRI) is often preferred over computed tomography (CT) because it does not subject the patient to radiation. Notably, patients may require sedation for MRI, and if this is not feasible, CT is an alternative modality.
How Should Abdominal Masses in the Pediatric Population Be Imaged?
Properly supervised US should be able to demonstrate greater than 90% of intraabdominal masses of the solid organs. Technically limiting factors, such as bowel gas, constipation, and patient agitation, can limit the utility of US. In those cases, rescanning after addressing the underlying limitation may be helpful.
In addition, US can help the radiologist plan proper additional imaging based on the appearance of the abnormality, its vascularity, and its relationship to other structures. In patients with negative US who are still highly likely to have underlying pathology, as well as patients with positive US who need further diagnostic imaging, imaging with CT with contrast or MRI without and with intravenous contrast can be the appropriate follow-up examination. Abdominal CT is fast and extensively available but has ionizing radiation, whereas MRI has improved soft tissue resolution but is often less available and may necessitate sedation because of the longer examination time. Other examinations may subsequently be needed for staging and evaluation for potential metastatic disease (such as chest CT or F-18 FluoroDeoxyGlucose Positron Emission Tomography-CT (F-18 FDG PET-CT)).
How Should a Differential Diagnosis Be Approached?
The combination of patient age, location of mass, and imaging appearance will narrow down the differential diagnosis significantly. Pathologies are often different in neonates/infants compared with older children, and although there may be mild overlap in age of presentation, individualized differential diagnoses for those age groups may be helpful.
Abdominal Masses in Neonates/Infants
In children younger than 1 year, the vast majority of abdominal masses are benign; in fact, approximately 80% are not even neoplastic but instead are developmental or inflammatory in etiology. When focusing on neonates (i.e., children <1 month old), roughly 75% of palpable abdominal masses arise from the genitourinary tract, including kidneys, adrenal glands, bladder, and reproductive system.
Renal Masses: Hydronephrosis, Cystic Renal Diseases, and Renal Neoplasms
Hydronephrosis
Hydronephrosis, the most common etiology of an abdominal mass in a neonate, often presents as a palpable flank mass. Major causes include uretero-pelvic junction (UPJ) obstruction, posterior urethral valves, UVJ obstruction, vesicoureteral reflux, neurogenic bladder, and prune belly. Because hydronephrosis is such a common entity, it is discussed in detail in a dedicated chapter in this book (see Chapter 8 , Imaging Approach to Urinary Tract Dilation).
Cystic Renal Diseases
Cystic renal diseases in the neonate include, but are not limited to, multicystic dysplastic kidney (MCDK) and autosomal dominant and autosomal recessive polycystic kidney disease. Whereas inherited cystic disease affects both kidneys, MCDK is a nonhereditary condition that is thought to develop secondary to a congenital complete obstruction at the ureteropelvic junction. Although MCDK is usually unilateral, patients with MCDK have a 20% to 50% chance of having other concurrent contralateral renal anomalies, for example, a UPJ obstruction or vesicoureteral reflux.
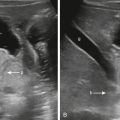
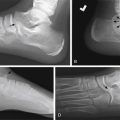
When cystic renal disease is suspected, US is the first-line imaging study. The typical sonographic appearance of a MCDK is a kidney replaced by multiple noncommunicating cysts and dysplastic-appearing intervening thin parenchyma ( Fig. 6.1 ); compensatory hypertrophy of the contralateral kidney will generally be seen, particularly as the infant grows. In some cases, it can be challenging to differentiate MCDK from unilateral hydronephrosis. In these cases, functional imaging with Tc-99m-MAG3 scintigraphy can be considered as a follow-up study to US. On scintigraphy, an MCDK will show no renal function, whereas a hydronephrotic kidney will usually show at least a small amount of renal function.

Autosomal dominant polycystic kidney disease will often not present until the child is older; however, macrocysts in a neonate should prompt a discussion of this entity with the referring clinician to evaluate family members because greater than 90% of these cases are inherited.
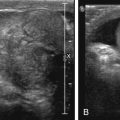
Autosomal recessive polycystic kidney disease will appear as markedly enlarged and diffusely echogenic kidneys as a result of tubular ectasia ( Fig. 6.2 ). Small renal cysts or areas of severe tubular ectasia may be seen; other times, only diffusely increased echogenicity is seen because the cystic spaces may be too small to resolve via US and instead will appear echogenic because of increased surface interfaces.

Renal Neoplasms
The most common renal neoplasm in children younger than 1 year is mesoblastic nephroma. Other renal neoplasms, such as Wilms tumor, are relatively rare in neonates and occur more frequently between the ages of 1 and 11 years. Wilms tumor will be discussed in greater depth later in this chapter.
Mesoblastic nephroma affects boys more often than girls. The mean age at diagnosis is 3 months. Ninety-five percent of cases are benign. Ten percent of patients will have a prenatal history of polyhydramnios. Patients typically come to medical attention when they are found to have a palpable abdominal mass; other clinical findings, such as hypertension and hypercalcemia, are rare. On US (either prenatal or postnatal) a mesoblastic nephroma will appear as a large, predominantly solid renal mass measuring from 5 to 30 cm in length. The mass is typically fairly homogeneous in echotexture. Generally speaking, hemorrhage and necrosis are absent except in the rare malignant form.
Once US has demonstrated a solid renal mass, further workup with contrast-enhanced MRI or CT is indicated ( Fig. 6.3 ). On cross-sectional imaging, mesoblastic nephroma is often noted splaying the renal calyces. On MRI, it is T1 hypointense and T2 hyperintense to adjacent renal parenchyma, a nonspecific appearance shared by many renal masses. It is crucial to evaluate for renal vein involvement. The radiologist should also look carefully for metastases to lymph nodes and/or lung (the latter is better demonstrated on CT), because these can be seen in the rare malignant form. Mesoblastic nephroma cannot be definitively differentiated from Wilms tumor on imaging, and nephrectomy is the standard of care. After nephrectomy, either US or MRI can be performed at intervals for postsurgical surveillance as clinically indicated.

Adrenal Masses
The most common solid malignant neoplasm in neonates/infants is neuroblastoma, a “small round blue cell tumor” arising from premature neuroectodermal cells and affecting 1 in 10,000 individuals. Although neuroblastoma can arise from sympathetic nervous stem cells anywhere in the body, most neuroblastomas in infants arise in the abdomen, usually from the adrenal medulla. The median age at presentation is 1 year 10 months, although some cases can be congenital. Symptoms can include diaphoresis, flushing, and diarrhea. Laboratory findings include high urine levels of catecholamine metabolites, such as homovanillic acid and vanillylmandelic acid. Pathologically, N-Myc gene amplification within tumor cells is associated with worse prognosis.
Imaging is crucial for accurate diagnosis and staging of neuroblastoma. Imaging evaluation typically begins with US, where neuroblastoma appears as a heterogeneous, predominantly solid mass often located superior to and separate from the kidney ( Fig. 6.4A ). Calcifications, cystic components, hemorrhage, and/or necrosis may be present. Neuroblastoma can be quite large at the time of diagnosis, often traversing the midline, surrounding vascular structures, and displacing the bowel. When the neuroblastoma is small, an important mimic is adrenal hemorrhage, which will not have internal vascularity on color Doppler and will decrease in size on follow-up ultrasounds, whereas neuroblastoma will have internal Doppler flow and typically stay the same size or enlarge on short-term follow-up. Another important mimic is infradiaphragmatic pulmonary sequestration (IPS), especially when the lesion is located in the left suprarenal fossa. Infradiaphragmatic sequestration is discussed in detail later in the Miscellaneous Abdominal Masses section.

Further cross-sectional imaging with MRI or CT is performed at the time of diagnosis for detection of imaging-defined risk factors (IDRFs). MRI is a sensitive modality for assessing the presence of local invasion, especially paraspinal/spinal involvement, and metastases ( Fig 6.4B ). Lymph node, osseous, and liver metastases are more common sites of involvement, whereas lung and brain metastases are generally seen only in advanced disease. MRI also demonstrates the relationship between the tumor and adjacent blood vessels, which may be in contact with, flattened by, or encased by tumor. On MRI, neuroblastoma will appear as a T1-hypointense, slightly T2-hyperintense mass with heterogeneous enhancement that encases, rather than displaces, adjacent structures. If MRI is not clinically feasible, CT can be performed as an alternative.
Necessary additional imaging at the time of diagnosis includes I-123 meta -iodobenzylguanidine (MIBG) scan, which can be used to identify primary uptake, as well as cortical bone, bone marrow, and soft tissue metastases. Important auxiliary imaging modalities for staging neuroblastoma include Technetium (99mTc) medronic acid (Tc-99m-MDP) bone scan, which can be used to evaluate for cortical bone metastases.
Ovarian and Uterine Masses
Ovarian masses in neonates are usually benign; most, in fact, are cysts. The first-line imaging modality for the evaluation of neonatal ovarian masses is US. Not only can US characterize an ovarian mass as either a simple cyst, complex cyst, or a solid mass, but it can also help determine whether there is associated ovarian torsion. Small simple cysts are safe to follow with US, whereas large simple cysts should be referred to a surgeon for possible excision because of the risk for torsion. Complex cysts can be treated with aspiration or excision. Excision is recommended for all solid masses.
The most common uterine abnormality that causes a palpable abdominal mass is hydrometrocolpos, which occurs when maternal hormones stimulate endometrial secretions that cannot be excreted because of the presence of an imperforate hymen, vaginal atresia, or persistent urogenital sinus. Hydrometrocolpos is generally easy to identify on US, with fluid distention of the vagina causing an ovoid hypoechoic structure, often with posteriorly layering proteinaceous debris ( Fig. 6.5 ).


Gastrointestinal Tract Masses
Hepatobiliary Masses: Infantile Hepatic Hemangiomas, Hepatoblastomas, Mesenchymal Hamartomas, and Choledochal Cysts
Infantile Hepatic Hemangioma
Infantile hepatic hemangiomas (IHHs) are the most frequently occurring benign liver mass in the neonatal age group. In the literature an important distinction has been made between congenital hemangiomas and infantile hemangiomas (IHs). IHs appear shortly after birth, stain positive for Glucose transporter 1 (GLUT-1), and rapidly proliferate before undergoing a slow spontaneous involution within the first decade of life.
Patients may present with a palpable right upper abdominal mass. Alternatively, they may present with symptoms of congestive heart failure (if the IHH causes significant arteriovenous shunting) or thrombocytopenia (if the IHH causes platelet sequestration). Because there is an association between IHH and cutaneous IH, patients with more than five cutaneous IHs will undergo an abdominal US to assess for the presence of IHH and other visceral hemangiomas. Unlike patients with hepatoblastomas, patients with IHH have serum alpha-fetoprotein (AFP) levels that, when age-adjusted using a pediatric nomogram, are normal.
The sonographic appearance of IHHs is often a hypoechoic mass, but IHHs may also be hyperechoic, or of mixed echogenicity. On grayscale images, large vessels may be seen coursing through the vessels, and with color Doppler, IHHs will demonstrate avid vascularity. They can be solitary or multifocal. If there is significant arteriovenous shunting secondary to an IHH, the celiac artery may be enlarged, with tapering of the abdominal aorta inferior to the celiac axis.
Contrast-enhanced MRI can be obtained for further workup if the diagnosis is still unclear. IHHs will be T1-hypointense and markedly T2-hyperintense. Large flow voids can be seen coursing through the lesion. While in their proliferative phase, IHHs demonstrate rapid and intense arterial phase enhancement ( Fig. 6.6 ). If imaged during involution, arterial enhancement may be variable and less robust.

Because the majority of IHHs involute spontaneously without the need for medical treatment (propranolol, steroids, interferon, or vincristine), embolization, or surgery, it is reasonable to follow them with serial USs.
Hepatoblastoma
Hepatoblastoma is the most common primary liver malignancy in children. Although the peak age of diagnosis is 1.5 to 2 years, this tumor is sometimes diagnosed in infants. Hepatoblastoma is relatively rare: whereas, say, neuroblastoma affects approximately 100 in 1 million individuals, hepatoblastoma affects only 13 in 1 million individuals. Risk factors include familial adenomatous polyposis, Beckwith-Wiedemann syndrome, and premature birth. Most of the time hepatoblastoma can be palpated on physical examination. Important laboratory findings include a serum AFP level that, when age-adjusted using a pediatric nomogram, is elevated; this can be used to help distinguish a hepatoblastoma from an IHH. In 10% of patients with hepatoblastoma, the expected increase in the serum AFP level is not seen—a poor prognostic factor.
As with the other neonatal/infant liver tumors, imaging workup begins with US, which will often show heterogeneous, mixed echogenicity mass or masses with a mean primary size of 10 cm. A heterogeneous appearance is due to variable internal hemorrhage, necrosis, and/or coarse calcification ( Fig. 6.7A ).

MRI or CT of the abdomen is performed for staging, using the PRETEXT (Pre-Treatment EXTent of tumor) system, describing extent of tumor in the liver, degree of vascular involvement, and extrahepatic and metastatic disease A CT scan of the chest is needed to evaluate for lung metastases, the most common site of metastatic involvement.
MRI of hepatoblastoma will demonstrate a heterogeneous T1-hypointense and T2-hyperintense hypoenhancing mass (see Fig. 6.7B–D ). Foci of T1 hyperintensity and T2 hypointensity may be present and compatible with hemorrhage and/or calcification.
Mesenchymal Hamartoma of the Liver
Mesenchymal hamartoma is the second most often encountered benign liver mass in the neonatal population. Mesenchymal hamartoma is a nonneoplastic mass consisting of disordered hepatic tissue, including hepatocytes, epithelium-lined bile ducts, connective stroma, and venous channels. They are almost never seen in patients older than 2 years and are sometimes diagnosed as early as the fetal period, during which time prenatal US may reveal a cystic-and-solid liver lesion that may cause mass effect on the lungs and adjacent blood vessels occasionally resulting in hydrops.
In the postnatal period, imaging workup also begins with US, on which mesenchymal hamartoma cystic-and-solid liver mass with a prominent cystic component is frequently divided into numerous locules by thin septations. Cysts may be macrocystic or microcystic. Microcystic lesions can mimic solid masses.
MRI can be useful in assisting with surgical planning. On MRI, mesenchymal hamartoma will appear as a well-demarcated liver mass whose solid component is T2 hypointense, while its cystic component(s) is strongly T2 hyperintense. The solid component shows variable enhancement on postcontrast images ( Fig. 6.8 ).