Abstract
White matter vanishing syndrome (WMS) is one of the most common hereditary leukoencephalopathies, affecting all ages, including newborns and adults. We report the case of a 6-year-old child admitted to the emergency department with sudden loss of consciousness, in whom herpetic encephalitis was suspected on the basis of recent herpes virus infection, and whose brain MRI showed Vanishing White Matter, subsequently confirmed by identification of the EIF2B5 gene mutation. Vanishing White Matter (VWM) syndrome, also known as infantile ataxia with central hypomyelination, is a leukodystrophy which is one of a wide range of rare genetic disorders primarily affecting the white matter of the central nervous system, caused by mutations in the c. This damage generally affects the deep areas of the brain, and does not spare the U-shaped fibers, a finding of great diagnostic interest for differentiating from other causes of leukodystrophies. MRI is a fundamental diagnostic test with good sensitivity for establishing the diagnosis because of a very good correlation between MRI aspects and mutations in the EIF2B1-5 genes. Guanabenz and Fosigotifatorsont represent 2 promising molecules for improving quality of life and prognosis in this population.
Introduction
The leukodystrophies constitute a large group of rare genetic diseases primarily affecting the white matter of the central nervous system, whose prognosis for patients has often been poor, due to the limited availability of effective curative treatments for the majority of leukodystrophies, as well as delayed diagnosis [ ]. White matter vanishing syndrome (WMS) is one of the most common hereditary leukoencephalopathies, affecting all ages, including newborns and adults. The disease is usually chronic and progressive [ ]. We report a case of leukodystrophy in a 6-year-old child whose onset was severe, mimicking herpetic encephalitis.
Case presentation
We report the case of a 6-year-old child, with no personal or family medical or surgical history, admitted to the emergency department with a sudden onset of consciousness 2 days prior to admission, associated with focal epileptic crises in the left upper limb. Interrogation with the parents revealed the notion of peribuccal skin lesions of a description consistent with a herpes virus infection 08 days previously, which had resolved spontaneously.
Clinical evaluation revealed an obnubilated child with a Glasgow Coma Scale (GCS) of 11/15, febrile to 38.9, hemodynamically and respiratorily stable, with no meningeal stiffness or purpuric skin lesions, and a completely normal clinical examination. Biological workup revealed a disturbed inflammatory panel with elevated white blood cells at 23638/mm3, C-RP at 73mg/l, normal hemoglobin at 17.3g/dl, normal metabolic panel with Na+ at 143mmol/l, Ca2+ at 97mg/l, creatinine at 8.4 mg/l, hepatic workup and hemostasis panel with no particularities. We performed an aerobic and anaerobic blood culture during the peak, which came up negative, with a sterile urine microbiological work-up.
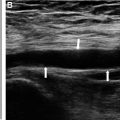
In view of this clinical context, the child’s age, the notion of herpetic cutaneous infection, the disturbance of consciousness and the localized epileptic crises, we suspected herpetic meningoencephalitis, and with this in mind, we carried out a cerebral MRI which first of all showed cerebral edema associated with bilaterally symmetrical diffuse signal abnormalities involving the periventricular supratentorial and subcortical white matter, with involvement of U-shaped fibers in T1 hyposignal ( Fig. 1 ), T2 and T2-Flair hypersignal ( Figs. 2-3 ), with hypersingal peripheral diffusion and Apparent diffusion coefficient (ADC) decrease ( Fig. 4 ), associated with atrophy of the trunk and splenium of the corpus callosum, and periventricular cystic remodeling, without enhancement after gadolinium injection, and without T2* hemorrhagic pattern ( Fig. 5 ).





We completed our workup with a cerebrospinal fluid study that returned normal and herpes polymerase Chain Reaction negative. In view of the diffuse, bilaterally symmetrical involvement of the supratentorial white matter, we retained the diagnosis of Vanishing White Matter, later confirmed by the identification of the EIF2B5 gene mutation. In terms of management, the patient was admitted to an intensive care unit (ICU), where secondary cerebral injury of systemic origin was first managed with paracetamol for fever, followed by a phenobarbital-based antiepileptic, and injectable Manitol for cerebral edema, with strict monitoring of the metabolic balance. The in-hospital evolution was favorable, with discharge from the ICU after 10 days without sequelae and with good clinical evolution. The patient and his family members are currently being monitored, given the increased risk of visual and cognitive disorders and cerebellar ataxia, a risk which has been fully explained to the parents.
Discussion
Vanishing White Matter (VWM) syndrome, also known as infantile ataxia with central hypomyelination, is a leukodystrophy which is one of a wide range of rare genetic disorders primarily affecting the white matter of the central nervous system [ ]. The condition mainly affects children, but recent reports have noted several cases diagnosed in adulthood [ ].
Genetically, and thanks to the advances in molecular biology that we made in 2001, it has been demonstrated that the genetic cause of the disease is caused by mutations in the EIF2B5 gene [ ]. This finding, which was found in a small group of patients from the eastern Netherlands with the same ancestor, and then the discovery of mutations in the EIF2B2 gene in the same population, led us to understand that possible mutations could concern any of the 5 genes coding for the eIF2B factor subunits [ ].
Subsequently, and with the introduction of whole-genome sequencing, over 120 mutations have been described in more than 250 patients, mainly involving the EIF2B5 and EIF2B2 genes, of which around 90% are missense mutations, most frequently affecting nonconserved amino acids, and a few recurrent mutations appear to be in paired cytosine/guanine (CpG) dinucleotides [ ].
On a macroscopic pathological examination, the brain generally appears normal, except in infants and young children, where flattening of the gyri can often be observed in conjunction with edema [ ]. In adults, enlargement of the lateral ventricles with subarachnoid spaces and cerebral atrophy may be observed. Macroscopically, however, the white matter is very pathological, with a pale grey or even cystic appearance [ ].
This damage generally affects the deep areas of the brain, and does not spare the U-shaped fibers, a finding of great diagnostic interest for differentiating from other causes of leukodystrophies [ ]. The areas most affected are mainly located in the deep cerebellar white matter, especially in the dentate nucleus, in the spinocerebellar tracts, the lateral and anterior cortico-spinal tracts and the anterolateral fasciculi containing the spinoreticular and spinothalamic tracts. This explains the symptomatology, which is dominated by cerebellar ataxia in the majority of cases [ ].
In immunohistochemical studies, myelin staining of the affected areas shows rarefaction of the myelin sheaths, with thinning and scattering by vacuoles of the remaining sheaths [ , ]. The presence of phagocytic cells characterized by orthochromatic cytoplasm and positive to Sudan black and Schiff’s periodic acid can be observed. Among the major observations of the immunohistochemical analysis is the significant degradation of myelin, with conservation of molecular weight and the presence of major myelin proteins that are mainly represented by oligodendrocyte myelin glycoprotein, cyclic nucleotide phosphodiesterase, and proteolysis protein [ ].
Clinically, the disease manifests as cerebellar ataxia and less marked spasticity. Cognition is often better preserved at the onset of the disease. Cranial pairs may be affected, with optic atrophy and impaired vision. Epileptic crises may occur during the course of the disease, but peripheral nervous system involvement is exceptional [ ]. The disease evolves in a chronic mode, but acute attacks can manifest as epileptic seizures, signs of intracranial hypertension with vomiting, hypotonia and, in certain situations, profound loss of consciousness, as described in our case. These acute attacks are usually secondary to acute stress, such as acute infection, fever >39º or minimal head trauma. In adults, the clinical presentation is generally less severe than in children, and is dominated by unexplained headaches and psychiatric symptoms [ ]. In affected women, a combination of leukoencephalopathy and primary amenorrhea or premature ovarian failure is possible, giving rise to the ovario-leukodystrophy syndrome [ ].
For biological markers, an increase in cerebrospinal fluid glycine levels can be observed, while a decrease in asialo-transferrin concentrations, but these observations lack sensitivity and specificity [ ].
Cerebral magnetic resonance imaging (MRI) plays a major role in the diagnostic algorithm for VWN syndrome, demonstrating diffuse, symmetrical white matter involvement that does not spare U-fibers in the form of signal abnormality in the form of hyposignal in T1 sequence and hypersignal T2 and T2 Flair [ , ]. In the advanced stages of the disease, MRI shows progressive rarefaction and cystic degeneration of the white matter. Diffusion sequences showed increased diffusion with hypersignal, with no enhancement of contrast after gadolinium injection. Proton spectroscopy showed a progressive reduction and eventual disappearance of all major metabolites, with glucose and lactate accumulating at similar concentrations to cerebrospinal fluid. It should be noted that there is a very good correlation between MRI aspects and mutations in the EIF2B1-5 genes, making MRI a fundamental diagnostic test with good sensitivity for establishing the diagnosis [ ]. However, there is no genotype-phenotype correlation, given the great phenotypic variability between patients with the same mutations, and from the same siblings, suggesting the impact of environmental and/or other genetic factors on the phenotype in a significant way.
Treatment is based first and foremost on general measures depending on the symptoms that predominate the clinical picture, such as management of spastic disorders and ataxia, treatment of epileptic seizures, prevention of acute stress triggers such as head trauma, infections, fever and malnutrition. In women with ovarian insufficiency, hormonal correction should be considered.
For specific treatment, there is the option of stem cell therapy. This therapy first proved effective in an animal model, with improved motor function and imaging abnormalities suggesting that transplantation of human glial cell populations may have positive therapeutic effects in patients with VWN [ , ].
For specific drugs, there are no drugs to date, but 2 clinical trials are on-going. The first clinical trial concerns Guanabenz, a molecule which has already proved its efficacy in an animal model and is currently being evaluated in humans (NTR7482). The second trial evaluates the efficacy and safety of Fosigotifator in children and adults with VWN syndrome (NCT05757141) [ ].
Conclusion
VWN syndrome remains a rare genetic disease, whose phenotypic presentation and prognosis are varied and variable from one individual to another. Guanabenz and Fosigotifatorsont represent 2 promising molecules for improving quality of life and prognosis in this population.
Patient consent
Written informed consent was obtained from the patient parents for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Competing Interests: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
1 Mohammed First University, Faculty of medicine and pharmacy, LAMCESM, Oujda, Morocco.
References
Related posts:
 A rare and life-threatening case of spontaneous hemopneumothorax presenting with severe hemodynamic instability
A rare and life-threatening case of spontaneous hemopneumothorax presenting with severe hemodynamic instability
 A very rare case of ileocolic and appendiceal intussusception with acute appendicitis
A very rare case of ileocolic and appendiceal intussusception with acute appendicitis
 Hepatic and extra-hepatic hydatid cysts: A case series of radiological and clinical insights
Hepatic and extra-hepatic hydatid cysts: A case series of radiological and clinical insights
 Mid-term follow-up of a fractured flow diverter in the internal carotid artery
Mid-term follow-up of a fractured flow diverter in the internal carotid artery
 An atypical multiple autoimmune syndrome: A case report including myocarditis
An atypical multiple autoimmune syndrome: A case report including myocarditis
 Successful ilio-femoral-popliteal venous aspiration thrombectomy using a 12 French system in a pediatric patient
Successful ilio-femoral-popliteal venous aspiration thrombectomy using a 12 French system in a pediatric patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


