KEY POINTS
Failure of closure of the neural tube during neurulation results in neural tube defects. Neurulation, both primary and secondary, are completed by approximately 32 post-ovulatory days.
Not all cases of neural tube defects are open lesions. Approximately 20% of spina bifida and 80% of cephaloceles are closed lesions. In this scenario the MSAFP is normal and the diagnosis is made at the time of the ultrasound examination.
Approximately 80 to 90% of children with Chiari II will develop hydrocephaly.
During the second and third trimesters the “classic” sonographic findings that aid in the diagnosis of spina bifida are the lemon and banana signs. Recently, three additional findings have been described: pointed lateral ventricles, beaked tectum, and interhemispheric cyst. In the first trimester, the newly proposed “intracranial translucency” may become an early sign of open spina bifida.
Anomalies of dorsal induction are those that result from failure or abnormal closure of the neural tube. Essentially these anomalies are better known as neural tube defects (NTDs). NTDs result from failure of the neural tube to close during primary neurulation. They are characterized by the presence of a cerebral, spinal, or combined cerebral/spinal defect or dysraphia.
The normal appearance of the fetal central nervous system (CNS) results from primary neurulation or dorsal induction; this in turn results in the formation of the brain and spinal cord exclusive of those segments caudal to the lumbar area. During primary neurulation, fusion of the neural fold occurs first in the dorsal region of the lower medulla at approximately 22 days after conception. Fusion does not proceed continuously in a caudal to rostral fashion as previously believed (the “zipper” theory).1 There are two fusion sites, and closure occurs bidirectional with the anterior neuropore closing before the rostral neuropore.2,3 Secondary neurulation, or caudal neural tube formation, occurs approximately between 26 and 32 postovulatory days and results in the formation of the lower sacral and coccygeal segments.4 It is during secondary neurulation that canalization occurs (see Chapter 1). Table 5–1 lists the defects that arise as a result of abnormal primary neurulation in decreasing order of severity.
In the United States, all pregnant women are routinely offered screening with maternal serum alpha-fetoprotein (MSAFP) for neural tube defects at 15 to 18 postmenstrual weeks. MSAFP is one of the components of the quad screen (alpha-fetoprotein, human chorionic gonadotropin, estriol, and inhibin-A), which is commonly used at present for screening of Down syndrome and open NTDs. Among low-risk women, MSAFP screening results in the detection of 80% to 90% of cases of fetal open NTDs.5 Limb et al6 published a study on the changes in prenatal detection and birth status of anencephaly between 1972 and 1990 in the Malformations Surveillance program of Brigham and Women’s Hospital in Boston. In the 1970s, half of the infants with anencephaly were born alive at an average gestational age of 35.6 weeks, and few were diagnosed prenatally; between 1988 and 1990, however, all affected fetuses were diagnosed either by prenatal ultrasonography or as a result of MSAFP, and the average age at delivery was 18 weeks.
MSAFP levels are expressed as multiples of the median (MoMs). An abnormal value is one that exceeds 2.5 MoMs. Elevated MSAFP levels are associated with NTDs, as well as a variety of other conditions (Table 5–2). In a retrospective study of 773 cases with elevated MSAFP, Reichler et al7 evaluated the percentage of fetal anomalies detected. They found that there was a progressive increase in the incidence of fetal anomalies as a direct function of the level of the MSAFP (Figure 5–1).
| Fetal Conditions |
| Multiple pregnancy |
| Intrauterine fetal demise |
| Wrong dates (i.e. pregnancy more advanced) |
| Ventral wall defects: omphalocele, gastroschisis |
| Renal: congenital nephrosis, bilateral renal agenesis, polycystic kidney or infantile |
| Intestinal atresia |
| Triploidy |
| Congenital skin disorders: epidermolysis bullosa or aplasia cutis |
| Teratoma: sacrococcygeal, pharyngeal |
| Congenital cystic adenomatoid malformation (congenital cystic adenomatoid malformation type III) |
| Turner syndrome with cystic hygroma |
| Oligohydramnios |
| Placental conditions |
| Hemangioma |
| Maternal Conditions |
| Maternal infection: parvovirus, cytomegalovirus, hepatitis |
| Maternal malignancy: hepatoma, ovarian teratoma |
| Abdominal pregnancy |
| Fetomaternal hemorrhage |
Figure 5–1.
Anomalies and oligohydramnios distribution as a function of elevated maternal serum alpha-fetoprotein (MSAFP). Other, subchorionic bleeding, intra-abdominal echogenicity, hydronephrosis, echogenic bowel, dilated kidney, heart defect; NTD, neural tube defect; VWD, ventral wall defect; Oligo, oligohydramnios; MoM, multiple of the median. (Reproduced, with permission, from Reichler et al. AJOG 1994;1071:1052.)7

NTDs can be categorized as open or closed, depending on whether or not they are covered by skin8 (Table 5–3). In an open NTD, the neural tissue is exposed or covered only by the thinnest of membranes; therefore, the lesion is directly in contact with the amniotic fluid. In these cases, the alpha-fetoprotein (AFP) molecule freely diffuses across the lesion, which results in an abnormally increased level in the amniotic fluid, hence in the maternal serum. Not all cases of NTDs are open lesions. For example, whereas all anencephaly are open defects, only 80% of spina bifida and 18% of cephaloceles are open NTDs.8 In a closed neural tube lesion, the defect is covered by skin or a thick membrane. In these cases, AFP cannot freely diffuse across the lesion into the amniotic fluid; therefore, the MSAFP level will be within the normal limits. Prenatal diagnosis in these cases will not be made on the basis of the serum or amniotic fluid AFP. An ultrasound (US) examination must therefore be performed in a timely fashion.
Acetylcholinesterase (AchE), unlike AFP, is not a normal component of the amniotic fluid. It is derived from neural tissue and is always seen in the amniotic fluid in the presence of an open NTD. Also, AchE may be present in cases of abdominal wall defects in which a nerve plexus is exposed to the amniotic fluid. It is essential to know that AchE is normally found in fetal blood and may therefore be found in amniotic fluid that at the time of amniocentesis has been contaminated with fetal blood.9,10
The occult dysraphic conditions affect the lower sacral and coccygeal areas of the spine. These lesions are covered by skin and may go on undetected, even after the birth of the infant. The important issue is that 4.1% of siblings of patients with these occult dysraphic conditions exhibit disorders of primary neurulation, such as meningocele and anencephaly.11,12
Anencephaly, exencephaly, acrania
Exencephaly is the absence of the calvarium and skin resulting in exposure of the brain. It appears to be the embryologic predecessor of anencephaly. Anencephaly is the complete absence of the calvarium, skin, meninges, and forebrain.
The reported incidence of NTDs of which anencephaly is the most common is about 1 case per 1000 live births worldwide.6,13,14,15 However, the incidence of NTDs is different among different patient populations. In the United States, the prevalence of NTDs is higher among Hispanic women than among non-Hispanic white or non-Hispanic black women.16,17 For example, in Puerto Rico, where most residents are Hispanic, the prevalence of NTDs is 8.68 per 10,000 live births, which is higher than in the mainland United States, which is 5.59 per 10,000.17
Anencephaly and related disorders are no longer theorized to be simple NTDs, but are complex developmental malformations that primarily affect the production of mesenchyme. This results in skeletal defects and imperfect fusion of the neural folds.18,19
The developmental sequence of events leading to anencephaly was first elucidated in experimental animals exposed to high doses of vitamin A. Subsequent studies in the human embryo have suggested that in humans it progresses in a similar fashion.19,20,21,22 The three phases in the development of anencephaly are (1) dysraphia or a failure of the neural groove to close in the rostral region (new evidence has suggested that the defect may occur as early as 18 to 20 postovulatory days, as a mesenchymal defect, far earlier than previously believed); (2) exencephaly, or exposure of a well-developed and differentiated brain outside the skull during the embryonic period; and (3) disintegration of the exposed brain during the fetal period, resulting in anencephaly4,18,20,21 (Table 5–4). It appears that early on during development the generation of the brain and the skull is relatively independent of each other and that the brain of anencephalic fetuses attains a high degree of differentiation before it disintegrates.21 Using prenatal sonography, the development of anencephaly from exencephaly has been observed and reported.23 This breakdown of the brain tissue has been corroborated by findings of primitive neuronal cells in the amniotic fluid.24
Exencephaly/anencephaly, as well as other NTDs, can be isolated or can be part of a malformation syndrome. Isolated NTDs, which account for the majority of cases, have a multifactorial origin involving both genetic and environmental factors.25 In these cases, one of the most important factors is the maternal serum folic acid levels. Maternal serum levels <200 μg/L have been associated with a significant risk of NTDs. Syndromes associated with NTDs can be the result of a chromosomal abnormality (eg, trisomy 13, 18, or 21) or a single gene disorder (eg, Meckel-Gruber syndrome).26 Teratogenic exposure results in a small number of cases of anencephaly and spina bifida.26 Maternal diabetes (pregestational) is associated with a 2- to 10-fold increase in malformations (eg, NTDs), and exposure to valproic acid and/or carbamazepine is associated with a 1% to 2% risk of spina bifida but not necessarily anencephaly.26 Other factors associated with NTDs are maternal obesity (patients with a body mass index [BMI] >29 kg/m2 having a 1.5- to 3.5-fold increase in risk) and hyperthermia (2-fold increase).26
Other malformations can also be seen in anencephalic fetuses. However, due to the severity and lethality of this malformation, searching for other malformations does not alter the management or the prognosis for the fetus. Among the other associated malformations are hypoplastic or anomalous folding of the ears; subcutaneous clefts of the nose, cleft lip, and/or palate; diaphragmatic hernia; omphalocele; limb and cardiac malformations; and hydronephrosis.27 Joo et al28 reported that in a series of 743 cases of NTDs, 385 had anencephaly, 307 had spina bifida, and 51 had encephalocele. They found that the most commonly associated anomaly with anencephaly is spina bifida (24%), a CNS anomaly; it can also be seen with cephaloceles (10%). Non-CNS malformations seen with anencephaly run the gamut of almost all organ systems, such as congenital heart defects in 6.5% (eg, atrial septal defect [ASD], ventricular septal defect [VSD], coarctation of the aorta, and univentriculat heart), gastrointestinal anomalies in 5.2% (eg, esophageal atresia, jejunal atresia, and intestinal malformation), urogenital anomalies in 3.1% (eg, hydronephrosis and polycystic kidneys), facial malformations in 19.2% (eg, hypo- and hypertelorism, proboscis, and cleft palate), limb anomalies in 4.1% (eg, clubfoot), and abdominal wall defects in 7.5% (eg, omphalocele, diaphragmatic hernia, and costal anomalies).28 Among 77 anencephalic fetuses who underwent karyotype, 97.4% had normal chromosomes, with one case each of trisomy 18 and 21.
Craniorachischisis refers to a defect in which the open cranial defect (anencephaly) is in continuity with the completely open spine (spinal dysraphism). In this defect total failure of neurulation has occurred and is believed to arise no later than 20 to 22 days after conception. Most of the fetuses affected with this extensive malformation are spontaneously aborted early in pregnancy.11 Craniorachischisisis presents in up to 10% of anencephalic fetuses.29 Sonography demonstrates the anencephaly and the extensive spinal dysraphism (Figure 5–2).
Figure 5–2.
Exencephaly at 17 postmenstrual weeks. (A) Coronal section demonstrating the eyes (lens) showing strabismus and the brain tissue without the calvarium. (B) The splayed upper part of the spinal column (arrow). (C) Coronal section highlighting the orbit with the lens pointing downward (strabismus). (D) A median section showing the flat profile (arrow). (E) and (F) Horizontal sections of the brain. No hyperechoic bony structure surrounding the brain tissue is seen. (G), (H), and (I) Pictures of the aborted specimen from the front, side, and back, respectively. Note the resemblance of the specimen to the respective sonographic pictures.

Polyhydramnios complicates up to 50% of anencephalic pregnancies, which usually develops during the second half of gestation. It is theorized that polyhydramnios results from decreased fetal swallowing.29,30,31,32
Anencephaly, like most NTDs, has a polygenic inheritance. In addition, several other factors, such as ethnicity, geographic location, and nutritional deficiency (eg, folate), may play significant roles in the occurrence of NTDs. The risk of an NTD increases significantly if there is a family history of an NTD. Deak et al15 reported on a data set of more than 1000 families affected by NTDs. They found both sex-influenced and maternal (imprinting) effects in the etiology of NTDs. Other factors associated with increased risk of NTDs are diabetes and exposure to valproic acid, thalidomide, and alcohol, as well as exposure to hyperthermia.33
Recurrence risk for NTDs is related to the family’s history. If one of the parents has an NTD, the risk to the offspring is as high as 4.5%.34 If a previous full sibling is affected, the recurrence risk is 4%; if two previous siblings are affected, this risk increases to as high as 10%. The recurrence risk among half-siblings is ~0.5% to 0.8%.15 The risk for second-degree relatives (eg, grandparents and grandchildren, uncles and aunts, nephews and nieces) is 0.5%, and for third degree (eg, first cousins), it is similar to that of the general population.15 In monozygotic twins, the concordance rate for NTDs is 7.7%; for dizygotic twins, it is 4%15 (Figure 5–3).

Folic acid can help prevent 50% to 70% of the cases of NTDs. Most studies suggest that folic acid works by correcting a nutritional deficiency of folic acid; however, exactly how it works is not known. Folates have two main physiologic effects: as a cofactor for the enzymes that synthesize DNA and RNA and for the conversion of homocysteine to methionine. The gene for 5, 10-methylenehydrofolate (MTHFR) catalyzes the conversion of MTHFR into 5-methyl-tetrahydrofolate, which is the major circulating form of folate. There are two mutations in the MTHFR gene that are associated with NTDs: the C677T and the A1298C mutations. Specifically, C677T has been linked to an increased risk of spina bifida and anencephaly. This mutation causes a mild enzymatic dysfunction that results in mild hemocystenemia in persons whose folate status is not optimal. Lower vitamin B12 levels during pregnancy have also been associated with increased risk of NTDs.1,35,36
A decrease in folate intake has been shown to be associated with an increased risk of NTDs.37,38,39 In view of this association, the U.S. Centers for Disease Control and Prevention (CDC) recommends that all women of childbearing age should consume 400 μg of folic acid per day for prevention of NTDs.40 However, public health campaigns have not had a significant impact on the prevalence of NTDs.26 Approximately 40% of women of childbearing age report taking folic acid, although racial and ethnic differences are noted, with Hispanic women who have the highest rate of NTDs reporting the lowest rate of consumption of folic acid.41 In 1998 the U.S. Food and Drug Administration (FDA) mandated that folic acid should be added to all grain products in the United States; since that time, many other countries have followed suit. Over the last 10 years since the fortification of cereal grain products was mandated, there has been a 26% decrease in NTDs in the United States.41 On the other hand, the amount of folic acid in these foods is small, and women of childbearing age still need to continue to supplement their diet with folic acid. Worldwide, as reported in 2007 by the CDC, the percentage of wheat flour fortification increased from 18% in 2004 to 27% in 2007.42 More recently, Bell and Oakley43 reported that the Flour Fortification Initiative now includes 67 countries that fortify wheat flour and 6 countries that fortify both wheat and maize (corn) flour; this has resulted globally in a 9% decrease of folic acid–preventable NTDs.
Recurrence risk can be significantly decreased with the use of periconceptional folic acid. For women who have had a prior child with an NTD, the recommended dose of folic acid is 4 mg daily started at least 1 month before conception and to be continued for the first 12 weeks of pregnancy.40 Doses of folic acid >1 mg must be taken under the supervision of a physician in order not to mask an underlying condition, such as pernicious anemia (vitamin B12 deficiency).
Anencephaly is a historically important malformation in the field of US, as it was the first malformation reported using transabdominal sonography in a fetus at 17 postmenstrual weeks.44 Almost 20 years later, it became the first malformation reported using transvaginal sonography (TVS) in a fetus at 11 weeks, 5 days (postmenstrual).45
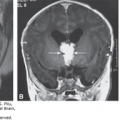
Using TVS, the integrity of the cranium can be assessed as early as the first trimester of pregnancy. This is because ossification of the fetal cranium begins and subsequently accelerates after 9 postmenstrual weeks.46,47 Abnormal mineralization of the cranial bones can be sonographically determined by the early second trimester by assessing the degree of echogenicity of the bone.48 Well-mineralized bone is highly echogenic. Absence of an echogenic outer border surrounding the fetal brain must raise the suspicion of the presence of exencephaly-anencephaly sequence (Figures 5–4 and 5–5). The visualization of echogenic “milky” amniotic fluid during the first or early second trimester is considered diagnostic for the presence of anencephaly (Figures 5–6 and 5–7).
Figure 5–4.
Anencephaly at 20 postmenstrual weeks. (A) Median section depicting a relatively normal profile of the fetal head. However, the skull is totally missing. (B) A coronal section of the face showing the fetal orbits with the lenses within. The arrow points to the two-vessel cord present in this fetus. (C) and (D) are two views of the hands of the fetus. Note that the hands are clenched with overlapping digits.

Figure 5–5.
The fetal head is seen in three different views in this fetus with exencephaly at 9 weeks, 5 days. (A), (B) Using two-dimensional (2D) sonography, the exposed brain is shown as disorganized and lacking any of the anatomical landmarks usually seen at this gestational age. (C) Three-dimensional (3D) reconstruction of this pathology. The typical appearance of the “Mickey Mouse”–shaped head is evident.

Figure 5–6.
A fetus with exencephaly-anencephaly sequence at 13 weeks, 3 days. (A), (B) The amniotic fluid (AF) appears echogenic when compared with the extraembryonic space (EES), which is anechoic; due to the disintegrating brain tissue, eventually no brain tissue will be seen, and the typical anencephalic appearance will be evident.


Exencephaly refers to a “transient” malformation in which the brain is exposed to the amniotic fluid. It is the second stage of the development of the clinically apparent anencephaly in humans.31,48,49,50,51,52,53 In exencephaly a relatively large amount of well-developed brain is present in the absence of a fetal cranium, with significant portions of the cranium missing, but there is preservation of the face and bones of the base of the skull. Preservation of the bones of the face and skull is also seen in anencephaly, although during the first trimester, specific structures, such as the ventricles and the choroid plexus, may be apparent (Figure 5–8). Usually, the first trimester exencephalic fetus has an apparently wide fetal head, with sonolucent spaces within the disintegrating brain.29 The outer shape of the head is bilobed; we, as well as other authors, have referred to this appearance as a “Mickey Mouse”–shaped head54 (see Figure 5–5). Exencephaly is rarely observed in human infants due to the disintegration of the exposed brain that occurs during intrauterine life. Most cases of exencephaly diagnosed in utero will have the typical anencephalic appearance at the time of delivery (Figure 5–9). Like anencephaly, exencephaly is a lethal malformation incompatible with postnatal life.
Figure 5–8.
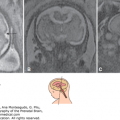
Merocrania in a fetus at 15 weeks, 6 days. Although the coronal plane (A) may produce the false impression of holoacrania, the medial plane (B) shows the presence of cranial bone. Note the relatively small anterior fontanelle with uncovered freely floating brain (arrows). (Courtesy of Gustavo Malinger.)

Figure 5–9.
Anencephaly at 31 postmenstrual weeks. (A)–(C) Subsequent coronal sections from the front to the back showing the widely spaced orbits (hypertelorism), the lens in the dislocated position (strabismus), and the scant tissue at the base of the skull. (D), (E) Median views showing the upper end of the vertebral column covered by a small amount of tissue (cerebrovasculosa). (F), (G) Lateral and posterior views of the specimen after birth.

The anencephalic fetus is easy to detect using sonography due to the severity of the malformation. This is especially true during the second and third trimesters of pregnancy. During the first trimester, the typical anencephalic phenotypic picture may not be sonographically apparent. Instead, an exencephalic fetus with an abnormally shaped head and some brain tissue may be imaged by sonography.
Anencephaly is characterized by the symmetric partial or total absence of the cranial vault above the orbits. In addition, a variable degree of disintegrating brain tissue may be present. The parts of the brain that are missing are the prosencephalon, the mesencephalon, and the rostral part of the rhombencephalon.18 In a sagittal view of a fetus with anencephaly, the profile of the chin, lips, nose, and orbits appears relatively normal, but superior to the area of the orbital ridges, the forehead and calvarium are obviously missing (Figure 5–10). A coronal view demonstrates the absence of the cranium above the prominent orbits with preservation of the base of the skull and facial features.55 The prominent, bulging eyes give the anencephalic fetus its typical “frog’s facies.” Several other abnormalities involving the eye and orbit of anencephalic fetuses have been described in pathologic specimens, such as coloboma, corneal dermoids, and anophthalmia.27 In addition, it has been reported that although the eyes of the anencephalic fetus may appear normal, often they have no connection to the brain centrally. Using sonography, we have noted the lenses of the anencephalic fetus to have an apparent strabismus, with both lenses located in the lower lateral aspect of the orbits. In most cases of anencephaly, a soft, spongy, red-colored vascular glial tissue simulating cerebral content is seen to protrude or to be exposed at the site of the defect. This tissue is commonly referred as the area cerebrovasculosa.
Figure 5–10.
A 3D reconstruction of the fetal face (using the surface rendering display) of a fetus with anencephaly at 21 weeks, 4 days. (A) Profile that demonstrates the typical bulging eyes, as well as the lack of calvarium above the fetal orbits. (B) The nose, lips, and chin appear normal, but the calvarium is missing just above the prominent fetal eyes.

Anencephaly can be further divided in two types, depending on the severity of the skull defect:
Holoacrania (Greek holos, “entire”), in which most or all of the calvarium is missing to the level of the foramen magnum. This is the typical anencephaly that is easily recognized by sonography.21,27,29 In addition, in holoacrania variable degrees of spinal rachischisis may be present.
Merocrania (Greek meros, “part”), in which there is a partial or incomplete median cranial defect with ectopia of the brain. The foramen magnum is not involved and no cervical lordosis is present. Merocrania may be confused with a cephalocele; however, it can be differentiated from it by the presence of cranial bones, sutures, and fontanelles and the absence of skin covering the ectopic brain21,27,29 (Figure 5–8).
Three-dimensional (3D) US is not essential to make the diagnosis of exencephaly-anencephaly sequence because two-dimensional (2D) US will provide all of the information needed for the diagnosis. However, it plays a key role in the counseling of the couple whose fetus has this anomaly as it will help the patient understand the severity of the anomaly. In addition to the reconstruction of the face (Figures 5–10 and 5–11), tomographic sections of the brain can be obtained to clearly define the defect (Figures 5–12 and 5–13).

Figure 5–12.
A typical-appearing fetus with exencephaly-anencephaly sequence is shown using the tomographic feature. Note that, compared with Figures 5–13 and 5–14, there is less visible brain tissue.


Anencephaly or exencephaly may result as a consequence of amniotic bands (see Figure 5–7). This nonrecurring cause of anencephaly can be differentiated from the anencephaly occurring as a result of failure of the neural tube to close, in that the cranial lesion is asymmetric, and multiple amputations of the fingers or toes, as well as defects of the abdominal wall, may also be present. The key sonographic finding in making this diagnosis of amniotic band syndrome is the presence of a band between the fetal defect and the placenta.53,56
Anencephaly is a lethal condition that more commonly affects female fetuses. It has been estimated that ~75% of fetuses with anencephaly are stillborn. Most infants born alive with anencephaly will die within the first 48 hours and the remaining within the first week of life,57,58 although rare prolonged cases of survival up to 14 months have been reported.59
Termination should be offered at the time of diagnosis, given the fact that this is a lethal anomaly. For those fetuses diagnosed at the time of delivery, compassionate and comforting care should be given. Most fetuses with anencephaly die within the first week of life; however, survival has been reported.60
None
Iniencephaly is a complex and lethal malformation that has three main features: a defect in the occiput involving the foramen magnum, retroflexion of the entire spine that forces the fetus to look upward with its occiput directed toward the lumbar region, and open spinal defects of variable degree.61,62,63,64,65,66
Iniencephaly is a rare malformation. The reported incidence of iniencephaly ranges from 1 to 6 per 10,000 births. Like anencephaly, most of the affected fetuses are female (90%).67
Iniencephaly, like anencephaly, results from a failure of fusion in the cervical and upper thoracic region of the upper spine. This lack of fusion secondarily results in a short neck and trunk, cervical and upper thoracic vertebral defects, defects of the thoracic cage, anterior spina bifida, diaphragmatic defects, and hypoplasia of the lung and/or heart.68 The malformation results from developmental arrest of the embryo no later than 24 days after conception. This results in persistence of the embryonic cervical retroflexion that leads to failure of the neural groove to close in the area of the cervical spine or the upper thorax.61,62,69 This defect most likely occurs only a few days later than anencephaly.
Iniencephaly has been divided into two types: iniencephalus clausus (or the closed type) and iniencephalus apertus (or the open type). In the latter, an occipital cephalocele protruding through the foramen magnum and occipital bone defect is present67 (Figures 5–14, 5–15, and 5–16).
Figure 5–14.
Iniencephaly at 12½ postmenstrual weeks. (A) Sonographic appearance in the median plane. Note the short body length due to the absence of the neck. Omphalocele was also present. L, liver. (B) The specimen from the side showing the shortened neck. (C) The dorsal view of the specimen showing the total spinal rachischisis. (D) Sagittal view of the specimen.


Figure 5–16.
A fetus with iniencephaly at 22 postmenstrual weeks. (A) Longitudinal median section. On the left of the picture, the outline of the head (H); on the right, the distorted vertebral column with several kyphoscoliotic deformations. Note the bulging brain tissue (occipital meningomyelocele, arrow). (B) Transverse section with the open vertebral column (rachischisis) is seen with the bulging brain tissue (arrow). (C), (D) The aborted specimen demonstrating the absence of a neck (iniencephaly) and rachischisis with meningomyelocele.

Iniencephaly, like all NTDs, has multifaceted causes, mostly a mix of genetic and environmental factors. Folic acid can decrease the recurrence rate.
Other malformations occur in up to 84% of fetuses with iniencephaly. Many of these associated malformations can only be diagnosed beyond the first trimester of the pregnancy. Among the associated anomalies, spina bifida is the most common, occurring in up to 50% of cases.70 Other anomalies are anencephaly, hydrocephaly, microcephaly, ventricular atresia, holoprosencephaly, polymicrogyria, agenesis of the cerebellar vermis, occipital encephalocele, cleft lip and palate, absence of the mandible, diaphragmatic hernia, thoracic cage deformities, cardiac malformation, urinary tract anomalies, omphalocele, clubfoot, and polyhydramnios.62,63,66,69,71,72
The recurrence risk of iniencephaly is 1.9%, which is similar to that quoted for most NTDs (see above).
Using TVS, iniencephaly has been diagnosed as early as 12.5 weeks’ gestation73 (Figure 5–14). On the median plane, the head appears large and held in retroflexion, the neck is not visualized, and the spine is usually lordotic (Figure 5–15). In cases of iniencephalus apertus, a posterior cephalocele is present in the occipital area (Figure 5–16). On transverse views, the open spinal defect is present. On axial sections, the head circumference may be several standard deviations below the mean, consistent with microcephaly. In addition, a size/dates discrepancy in a well-dated pregnancy may be the first sign of an iniencephalic fetus.
The main differential diagnosis is the Klippel-Feil syndrome (KFS). In the typical clinical scenario, there is a triad of findings consisting of a short neck, low posterior hairline, and limited neck movement, although <50% of patients demonstrate all three clinical features.74 Findings detected by prenatal sonography are cervical vertebral anomalies, short neck, low-set ears, and facial asymmetry.75 Other conditions that should be considered are Jarcho-Levin syndrome, Gorlin syndrome, anencephaly with raschischis, and cervical encephalocele.
Iniencephaly, especially the open type, is essentially a lethal anomaly, with most of the fetuses with this condition being stillborn or dying shortly after birth. However, long-term survival has been reported of up to 2 years of age.72
Termination should be offered at the time of diagnosis, given the fact that this is a lethal anomaly. For those fetuses diagnosed at the time of delivery, compassionate and comforting care should be given. Dystocia of labor may occur due to the abnormal head position.
Cranium bifidum, encephalocele
Cephaloceles are cranial defects, along bony sutures, in which there is a herniation of the brain and/or meninges. Sporadic or nonsyndromic cephaloceles account for ~5% of all NTDs.76 When the cephalocele sac contains brain tissue, it is termed an encephalocele; if only cerebrospinal fluid is present (CSF), it is termed a meningocele.
The reported incidence ranges from 1 per 3500 to 1 per 5000 live births.77 It is estimated that meningoceles are approximately 10 times less common than encephalocele.78 Cephalocele may involve the occipital, frontal, temporal, and parietal regions of the fetal head (Table 5–5). The occurrence of the different types of cephaloceles shows geographical variation. In Europe and North America, 66% to 89% of all cephaloceles are occipital, with the balance being equally distributed among frontal and parietal cephaloceles. In Thailand and countries of southern Asia, the frontal (sincipital) location is more common than the occipital.14,32,55,78,79,80,81,82
Occipital cephaloceles occur more commonly in female than in male fetuses, in contrast to parietal and sincipital cephaloceles, which are more prevalent in males (Figures 5–17, 5–18, 5–19, 5–20, 5–21, 5–22, and 5–23).78 The development of most severe cephaloceles takes place no later than 26 days after conception, when the anterior neural tube closes.11
Figure 5–17.
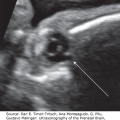
Posterior encephalocele at 12 weeks, 6 days. (A) Series of parallel sagittal sections as viewed using the tomographic imaging display showing the brain tissue extending into the amniotic sac (arrow). (B) Picture generated using surface rendering showing the head with the posterior encephalocele from a lateral view.

Figure 5–18.
Using transabdominal sonography, a large posterior encephalocele is seen in this fetus at 19 postmenstrual weeks. (A) An axial section displays the large cephalocele sac. Brain tissue is seen extending into the sac. (B) Sagittal section displays the encephalocele. (C) A 3D reconstruction of the posterior encephalocele.

Figure 5–19.
A small posterior encephalocele arising from the posterior aspect of the sagittal suture is seen using 3D transvaginal sonography in a patient at 21 weeks, 4 days, referred to our unit for evaluation of borderline ventriculomegaly. (A) The volume is displayed using the three orthogonal planes (Box A: sagittal; Box B: coronal; Box C: axial) the rendered imaged is seen in the lower right box. (B), (C) Volume displayed using tomographic imaging. In B the image is zoomed, and the small cranial defect (arrow) is seen in serial sagittal sections; in C in serial coronal sections. The herniated brain, B, is seen within the cephalocele sac.

Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


