specificity; therefore, they will cross-react with nuclei from different sources. The primary sources for study of these antibodies are patients with SLE and related systemic rheumatic diseases. Many studies have centered on defining the specificity of these antibodies and have contributed extensively to our understanding of their immunopathologic role in connective tissue disorders. It is worthwhile to state that 100% of patients with SLE are ANA positive; the concept of ANA-negative lupus is no longer valid. Furthermore, autoantibodies against small nuclear ribonucleoprotein (snRNP), also known as anti-Smith antibodies, were found to be highly specific for SLE, although they are present in only 15% to 30% of SLE patients. They are present more frequently (about 60%) in young black women with SLE. The immunobiology of lupus is well beyond the scope of this text, but we have to emphasize that despite exhaustive studies of tolerance and the existence of mouse model of lupus, there have not been any clinically significant advances in the specific treatment of SLE in nearly two decades.
Table 8.1 CLINICAL AND IMAGING HALLMARKS OF CONNECTIVE TISSUE ARTHRITIDES (ARTHROPATHIES) | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
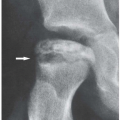
and/or accompanied by articular erosions (Fig. 8.3). Some patients present with sclerosis of the distal phalanges (acral sclerosis) (Fig. 8.4) or with resorption of the terminal tufts (acroosteolysis). Osteonecrosis, which is frequently seen, has been attributed to complications of treatment with corticosteroids (Fig. 8.5). However, current investigations suggest the vital role of the inflammatory process (vasculitis) in the development of this complication.
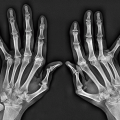
 Figure 8.1 ▪ Systemic lupus erythematosus. A: Typical appearance of the thumb in a 43-year-old woman with SLE. Note subluxations in the first carpometacarpal and metacarpophalangeal joints without articular erosions. B: In another patient, a 32-year-old woman, the oblique radiograph of her left hand shows dislocations at the first carpometacarpal joint and distal interphalangeal joint of the index finger (arrows) and subluxations in the metacarpophalangeal joints of the index and middle fingers associated with swan neck deformities (open arrows). |
 Figure 8.2 ▪ Systemic lupus erythematosus. A: Lateral radiograph of both hands of a 42-year-old woman with documented SLE for the past 4 years demonstrates flexion deformities in the metacarpophalangeal joints. B: On the dorsovolar projection, the flexion deformities have been corrected by the pressure of the hands against the radiographic cassette. |
 Figure 8.3 ▪ Systemic lupus erythematosus. A: A 62-year-old woman presented with a 15-year history of SLE. Dorsovolar radiograph of both hands shows severe deformities, subluxations, and articular erosions. Note the advanced osteoporosis secondary to disuse of the extremities and treatment with corticosteroids. B: In another patient, a 51-year-old woman, note flexion contractions, subluxations, and dislocations in the several joints of the right hand. |
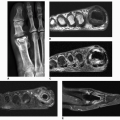
 Figure 8.4 ▪ Systemic lupus erythematosus. Dorsovolar radiograph of the hand of a 29-year-old woman demonstrates sclerosis of the distal phalanges (acral sclerosis). Similar sclerotic changes are also occasionally seen in rheumatoid arthritis and scleroderma. |
anticentromere. Recently, researchers have identified a new genetic link to systemic form of scleroderma, a susceptibility locus involving the genome CD247 (that encodes the T-cell receptor zeta subunit modulating T-cell activation), in addition to previously known genes MHC, IRF5, and STAT4 (that encodes a regulatory protein important to immune system). However, these linkages are weak and do not provide a clear-cut genetic tool that is useful for patients. Clinically, many patients develop joint involvement, which is manifesting as arthralgia and arthropathy leading to flexion contractions of the fingers. Most patients have the so-called CREST syndrome, which refers to the coexistence of calcinosis, Raynaud phenomenon (episodes of intermittent pallor of the fingers and toes on exposure to cold, secondary to vasoconstriction of the small blood vessels), esophageal abnormalities (dilatation and hypoperistalsis), sclerodactyly, and telangiectasia; 30% to 40% of patients have a positive serologic test for rheumatoid factor and a positive antinuclear antibody (ANA) test.
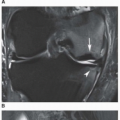
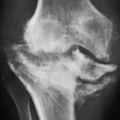
 Figure 8.5 ▪ SLE complicated by osteonecrosis. Oblique radiograph (A) and lateral conventional tomogram (B) of the ankle demonstrate osteonecrosis of the talus in a 26-year-old woman with lupus who was treated with massive doses of steroids. C: Coronal T2-weighted MRI in an 18-year-old woman demonstrates a focal area of osteonecrosis in the left femoral head. |
calcifications within the upper and lower limbs can occasionally be quite prominent (see Figs. 8.8 and 8.9B, C). Corroborative findings are seen in the gastrointestinal tract, where dilatation of the esophagus and small bowel, together with a pseudo-obstruction pattern, is characteristic (Fig. 8.11). Pseudodiverticula in the colon are also commonly seen.
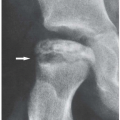
 Figure 8.6 ▪ Scleroderma. A: A 24-year-old woman presented with atrophy of the soft tissues at the distal phalanges of the index, middle, and ring fingers (arrows). B: A radiograph of the fingers of the left hand of a 36-year-old woman shows atrophy of the soft tissue at the tip of the index and middle fingers (arrows) and early resorption of the terminal tufts (arrowheads). Compare with normal ring finger.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|