The symptoms associated with slow viral or prion diseases of the central nervous system tend to have multiple neurologic symptoms, and different patients may present with different symptoms. This review discusses the most common slow virus infections and their imaging findings.
Slow virus diseases may be caused by conventional viruses or by unconventional viruses (atypical viruses or agents). The term slow virus infections refers to the tempo of the disease rather than to the growth rate of the virus. Such diseases have a prolonged incubation period (which can be months or years) and a protracted progressive clinical course. The symptoms associated with slow viral or prion diseases of the central nervous system (CNS) tend to have multiple neurologic symptoms, and different patients may present with different symptoms. This review discusses the most common slow virus infections and their imaging findings.
Viruses
Progressive multifocal leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is a subacute opportunistic infection caused by the unenveloped DNA virus JC Polyomavirus (JCV). By the age of 65 years, 50% to 70% of individuals have antibodies to the virus . After entering, the virus persists in the renal tubular epithelial cells. With reactivation of latent JCV in the kidneys and subsequent viremia, the JCV enters the brain and causes PML.
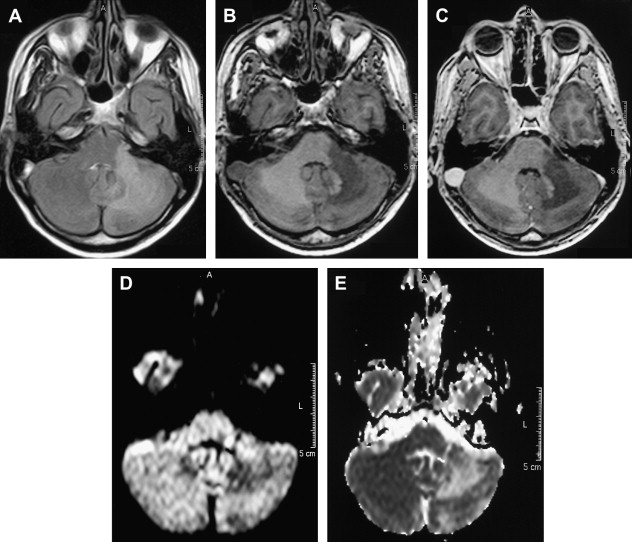
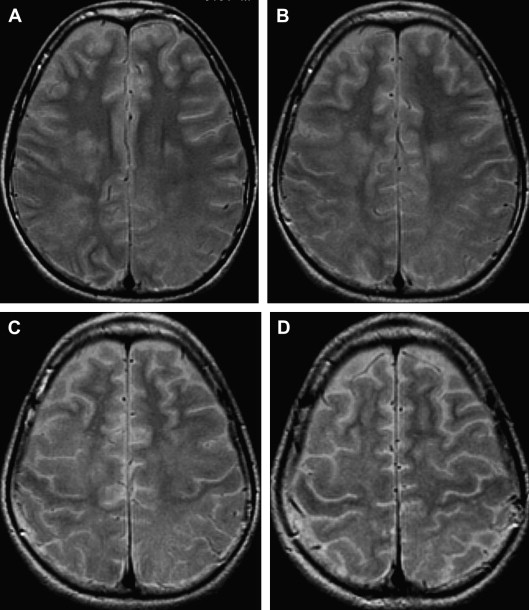
The incidence of PML has greatly increased as a result of the AIDS epidemic, and 0.7% to 11% of patients who have HIV develop PML during the course of their illness . The histopathologic hallmark of PML is demyelination with enlarged oligodendroglial nuclei and bizarre astrocytes. The disease is usually multifocal, and the lesions may occur in any location in the white matter, most often in the parieto-occipital region. Thalamic lesions are frequently present, and the cerebellum and brain stem can also be involved. Posterior fossa lesions are usually present simultaneously with supratentorial lesions, and, in rare cases, PML may be limited to the posterior fossa ( Fig. 1 ) . Although there are no significant pathologic differences between patients who have and do not have AIDS, in the AIDS population, more extensive disease with a necrotizing character was observed .
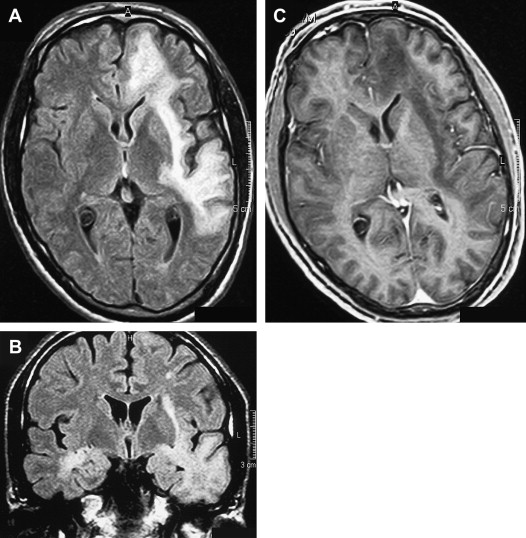
The findings on MR imaging correlate well with macroscopic changes. On T2-weighted MR imaging, PML lesions are patchy, scalloped, high signal intensity lesions located in the white matter, with extension along the white fibers ( Fig. 2 ) . Subcortical arcuate fibers are involved, mass effect is mild or absent, and peripheral faint enhancement is a rare feature . On T1-weighted images, the PML lesions have low signal, in contrast to isointense HIV-associated lesions (see Fig. 1 ) .
A recent study demonstrated the existence of a significant difference between the magnetization transfer characteristics of PML lesions and white matter lesions attributable to HIV-associated dementia (HAD) in patients who have AIDS . In that study, the mean magnetization transfer ratio (MTR) value of PML lesions was 26.1%, compared with 47.9% in HAD lesions . The reported difference in MTR between PML and HIV-associated lesions most likely reflects differences in pathophysiology and supports the fact that the high MTR of normal white matter is attributable to the presence of myelinated axons. MR imaging, with the magnetization transfer saturation technique as a noninvasive tool, can be used to differentiate between PML and HIV-related lesions in patients who have AIDS. A central area of marked hypointensity on T1-weighted MR imaging and hyperintensity on T2-weighted MR imaging was observed at follow-up, suggesting necrotic changes . These findings are consistent with the low MTR values observed in PML lesions. Prominent necrotic changes in PML lesions in patients who have AIDS were also found in neuropathologic studies .
Proton ( 1 H) magnetic resonance spectroscopy (MRS) was also used to image PML lesions, and the spectra were compared with those in normal brain parenchyma. The spectra of PML lesions were characterized by significantly reduced N-acetylaspartate (NAA), lactate presence, and increased choline and lipids . A decrease in NAA is the result of axonal loss, and the presence of lactate is related to cellular hypoxia. The increase in choline and lipid may reflect an accumulation of myelin breakdown products.
Thallium-201 ( 201 TI) brain single-photon emission computed tomography (SPECT) has also been used in patients who have AIDS for differentiation of malignant lesions from lesions of infectious origin. In one study, combined gallium and thallium scans were performed on patients who had AIDS with focal brain lesions . Patients who had PML had two different patterns: patients with positive gallium scans and negative thallium scans and a second group with negative thallium and gallium scans. Some PML cases can thus be mistaken for malignant lesions. Demyelination and destruction explain negative thallium and gallium scans. Positive gallium and negative thallium scans may be the result of coexisting pathologic changes.
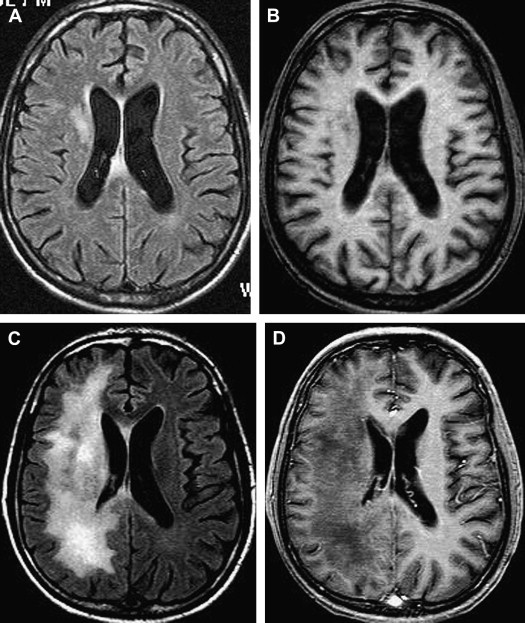
Without treatment, the prognosis for PML is usually poor, with death occurring after 2.5 to 4 months. Only a small number of cases have been reported to have a more benign clinical course . In one large prospective study of 48 patients who had AIDS with proved PML, except for mass effect, which was associated with shorter survival, no magnetic resonance abnormality significantly correlated with patient survival . Although, at present, there is no efficacious therapy for PML, recent studies have shown clinical and radiologic improvement in patients who had PML and underwent highly active antiretroviral therapy (HAART) . In patients with prolonged survival regression or stabilization, MR imaging findings paralleled suppression of virus replication and immune response recovery ( Fig. 3 ) . In one study, initial worsening of the MR imaging findings, with development of temporary contrast enhancement, mass effect, and edema, was observed in patients who were long-term survivors . This was probably a result of a posttreatment inflammatory reaction attributable to the immune reconstitutive effect. Atrophic changes and increased hypointensity on T1-weighted imaging with concomitant low signal on fluid-attenuated inversion recovery (FLAIR) images in those patients represent leukomalacia and burnt-out PML lesions (see Fig. 3 ). In patients with no response, progressive disease can be recognized by increasingly high signal on T2-weighted and FLAIR images and increasingly low signal on T1-weighted images as a result of demyelination. Approximately half of the patients who have AIDS with PML do not experience benefit from HAART. Tantisiriwat and colleagues showed that PML can also develop in patients who have AIDS and are undergoing HAART, making the interpretation of the imaging findings even more challenging.
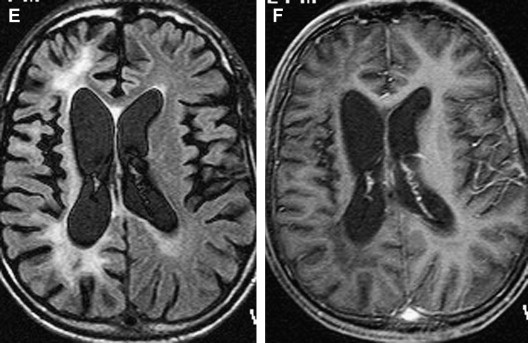
Until 2005, PML was exclusively reported in patients who had AIDS or in immunosuppressed patients who had malignant diseases. Two cases of PML have been described recently in patients who had multiple sclerosis (MS) and were treated with the α-integrin inhibitor natalizumab . Natalizumab was introduced in 2004 for the treatment of relapsing-remitting MS. One possible explanation is a natalizumab-triggered reactivation of latent JCV in the kidneys . After being withdrawn from the market for awhile, the agent was reintroduced in June 2006 under a careful risk management program (Tysabri Outreach: Unified Commitment to Health Prescribing [TOUCH] program) to allow assessment of the incidence and risk factors for PML in natalizumab-treated patients . In patients who have MS and are taking natalizumab, the occurrence of new or worsening symptoms mandates the need for a standard cranial MR imaging scan. A suggested brain MR imaging protocol for the detection of PML includes proton density, T2-weighted, FLAIR, and T1-weighted MR imaging with and without gadolinium .
Furthermore, recent findings on viremia in healthy individuals raised the possibility of infection persistence at a basal level and reactivation in immune dysfunction . The possibility of JCV persistence in the brain with reactivation from that site has also been discussed among researchers . Important questions on pathogenesis, trafficking, and reactivation of JCV need to be answered in the future.
Subacute sclerosing panencephalitis
Subacute sclerosing panencephalitis (SSPE) is a progressive fatal neurologic disorder of childhood and early adolescence. It is caused by defective measles virus and develops after an asymptomatic period of 6 to 8 years. The measles virus (MV) is a member of the Morbilliviruses. Histopathologic findings include astrogliosis, neuronal loss, demyelination, neurofibrillary tangles, and infiltration of inflammatory cells .
The prevalence of SSPE is higher in countries with inadequate immunization systems. India is reported to have a 10-fold higher prevalence compared with most other countries . SSPE usually begins with the insidious onset of mental deterioration and motor dysfunction, with progression to a vegetative state and death over a period of 1 to 3 years . It is diagnosed based on clinical findings, electroencephalography (EEG), and cerebrospinal fluid (CSF) features.
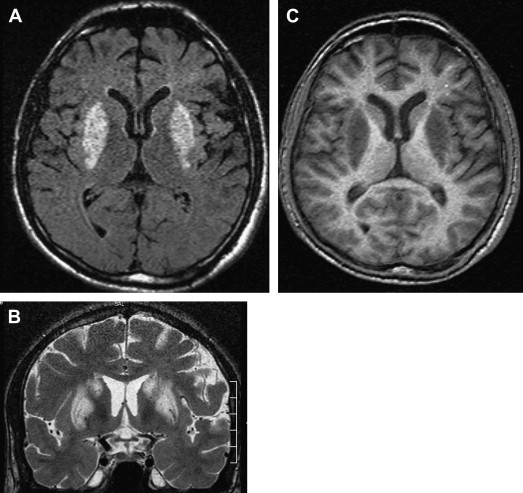
CT findings are generally normal in the early stages of SSPE. As the disease progresses, atrophic changes and hypodensities of white matter can be observed as indicators of demyelinating areas . MR imaging is a superior method for detecting white matter abnormalities in SSPE . MR imaging studies on SSPE have reported characteristic bilateral, asymmetric, hyperintense lesions in the parietal and temporal lobes in the acute stage . With time, lesions become more prominent, and the periventricular white matter, the corpus callosum, and the basal ganglia can be involved ( Fig 4 ). Encephalomalacia and atrophy develop in later stages . A recent study describing imaging findings in six patients who had acute measles encephalitis reported the presence of striatal necrosis, hemorrhagic parenchymal lesions, and parenchymal and leptomeningeal enhancement after administration of intravenous contrast medium .
In one report, conventional MR imaging and MRS were performed in a case of early-stage (stage II) SSPE in an 8-year-old child . MRS demonstrated increased myoinositol (MI)/creatine (Cr) and choline/Cr ratios, and a decreased NAA/Cr ratio. Similar results have been reported when using single-voxel MRS and chemical shift imaging in late-stage SSPE . In early-stage SSPE, the NAA/Cr ratio was found to be normal.
The most commonly involved areas in SSPE are the periventricular and subcortical white matter (see Fig 4 ). The basal ganglia, cerebellum, spinal cord, and corpus callosum are less commonly involved ( Fig 5 ). Brain stem involvement is rare and is usually accompanied by other intracranial lesions. In one article, the investigators reported two cases of SSPE associated with brain stem involvement .
Diffusion tensor imaging (DTI) with fractional anisotropy (FA) and mean diffusivity (MD) values in the periventricular white matter, corpus callosum, and posterior limb of the internal capsule were measured in 21 patients who had stage II SSPE and 10 age- and gender-matched healthy controls . In that study, patients with normal findings on conventional imaging showed significantly lower FA values than did healthy controls. Abnormal- and normal-appearing white matter on T2-weighted images showed significantly decreased FA values in all regions compared with those in healthy controls. MD values were significantly increased in the periventricular white matter region of the frontal and parieto-occipital lobes in patients with normal and abnormal findings on conventional imaging compared with those in healthy controls .
It remains to be proved in larger studies whether DTI is a useful method in the early detection of SSPE and whether it may be helpful in treatment planning for these patients.
The fulminating form of SSPE is an extremely rare condition. Imaging findings usually do not correlate with clinical staging .
A small number of patients have prolonged survival from 3 to 13 years . The role of the immunologic status of the patient, wild strains, and the altered virulence of the virus in the pathogenesis of SSPE are the subjects of future investigations.
Progressive rubella panencephalitis
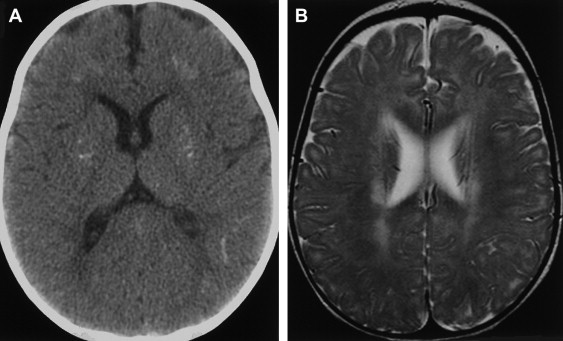
Progressive rubella panencephalitis (PRP) is caused by the rubella virus. It was first reported in 1974, with few reports in the literature since then. All reported patients were male, and most had signs of congenital rubella syndrome. Clinical features of PRP resemble those of SSPE, but the age at onset is much older and the clinical course is more benign. The main neurologic features of PRP include dementia, cerebellar ataxia, and seizures. Increases in antirubella antibody titer and IgG are found in the CSF, and diffuse atrophy of the brain with ventricular dilatation may be found on MR imaging ( Fig. 6 ) .
Rabies
Rabies (also known as hydrophobia or aquifuga) is an acute CNS infection caused by the rabies virus or related members of the genus Lyssavirus , family Rhabdoviridae. The term rabies is derived from the old Indian word rabh , meaning to make violent . The virus is usually transmitted through a dog bite. Serious precautions are needed when traveling in countries in which rabies is endemic, and any dog bite in such a country should be considered potentially dangerous. If no postexposure vaccination is given, the risk for developing rabies after the bite of a rabid dog is approximately 50%, depending on the location and severity of the bite . Inflammation and demyelination of the spinal nerve roots and peripheral nerve are characteristic findings in paralytic rabies . The clinical picture of rabies encephalitis is unmistakable. It starts with malaise, fever, and paraesthesia, followed by classic neurologic symptoms of agitation, hydrophobia, aerophobia, hypersalivation, and seizures . Approximately 20% of rabies occurs in its paralytic form. Paralytic rabies lacks the classic symptoms and can be confused with Guillain-Barré syndrome (GBS) and related disorders and other treatable autoimmune diseases of the peripheral nerves. Only a few reports on MR imaging findings in rabies have been found in the literature . In human rabies of both forms, the spinal cord gray matter and the anterior horn cell were involved (detected as high signal intensities on T2-weighted images). The lateral and posterior columns were also affected, however . Enhancement along the brachial plexus of the bitten arm was noted in one patient. Enhancement with gadolinium-based contrast material was seen at the hypothalami, brain stem nuclei, spinal cord gray matter, and intradural cervical nerve roots only when the patient became comatose . In one study, extensive magnetic resonance abnormalities in the brain stem and hippocampus in a patient who had rabies encephalitis that had been proved at necropsy were reported . Hyperintensities in the globus pallidi, putamen, and thalami bilaterally on T1- and T2-weighted images in a case of rabies encephalitis diagnosed antemortem have also been described . High signal intensity abnormalities seen on T2-weighted MR imaging bilaterally in the hippocampus, thalamus, basal ganglia, and dorsal aspect of brain stem reflect the pathologic studies of rabies that have shown maximal concentration of Negri bodies and antirabies antigen, as revealed by immunohistochemistry .
Recently, rabies encephalomyelitis was reported in three patients who received solid organ transplants from a common donor . Serologic testing performed postmortem confirmed rabies virus infection in the common donor.
Prions
Prion diseases refer to subacute spongiform encephalopathies with infectious and genetic components. Prions replicate without provoking an antibody response, and they are resistant to conventional inactivation techniques for infectious agents. The prion protein is an abnormal isoform of a normal host-encoded protein and is located on the short arm of chromosome 20. The isoform abnormal form accumulates in prion diseases in the CNS. Creutzfeldt-Jakob disease (CJD), kuru, and Gerstmann-Straüssler syndrome are prion diseases in human beings. Scrapie, transmissible mink encephalopathy, chronic wasting disease of mule deer and elk, and the recently discovered bovine spongiform encephalopathy (BSE) are similar diseases found in animals.
Creutzfeldt-Jacob disease
CJD is a rare and invariably fatal disorder of the CNS caused by a prion. The disease is diagnosed by the detection of an abnormal form of the human prion protein PrP Sc in the brain . A brain biopsy or autopsy is required to confirm the diagnosis , but laboratory tests that detect the 14-3-3 protein in the CSF, an elevated concentration of neuron-specific enolase (NSE), and periodic sharp wave complexes on EEG, along with clinical signs, allow for a probable diagnosis while the patient is still alive . The incubation period of CJD ranges from 3 to 22 years, with a subacute onset and progressive dementia. In general, the disease affects the elderly population, with a peak incidence between 60 and 64 years, but it has been demonstrated in young individuals as well , and at least certain forms, such as sporadic CJD, seem to affect women more commonly then men (2:1 ratio) . The disease is characterized by rapidly progressive dementia; short-term illness; and focal involvement of the cerebral cortex, basal ganglia, cerebellum, brain stem, and spinal cord. The patients may develop myoclonic movements and pyramidal (Babinski sign) and extrapyramidal symptoms (rigor, akinesia, and choreatic movements) . The infectious agent is present in the brain, spinal cord, eyes, lungs, lymph nodes, kidneys, spleen, liver, and CSF but not in other body fluids.
CJD can be divided into four types: the familial form (inherited form; fCJD), the iatrogenic form (from contaminated allograft tissue and surgical instruments), the variant form (vCJD), and the sporadic form (sCJD).
The sporadic form is the most common type and accounts for up to 85% of the clinical cases. Different clinical and pathologic subtypes of cCJD have been linked to polymorphism in methionine or valine status at codon 129 PRNP . Genotyping of the PRNP codon 129 has resulted in several different molecular subtypes, with the MM genotype (methionine homozygosity) being the most frequently occurring genotype, followed by the MV and VV genotypes in sCJD , which can then, on the basis of phenotype, be classified as type 1 or type 2. Depending on the subtypes, age at onset of illness, disease duration, and clinical symptoms, MR imaging findings might vary. Such patients often present with cognitive decline, followed by the development of a variety of neurologic symptoms, however. Cerebellar symptoms are a common feature of the disease and are often present at the time of onset or throughout the illness, accompanying other general global symptoms . Isolated cerebellar symptoms as early manifestations appear far less commonly and have been demonstrated in only 5% of cases in a recent study .
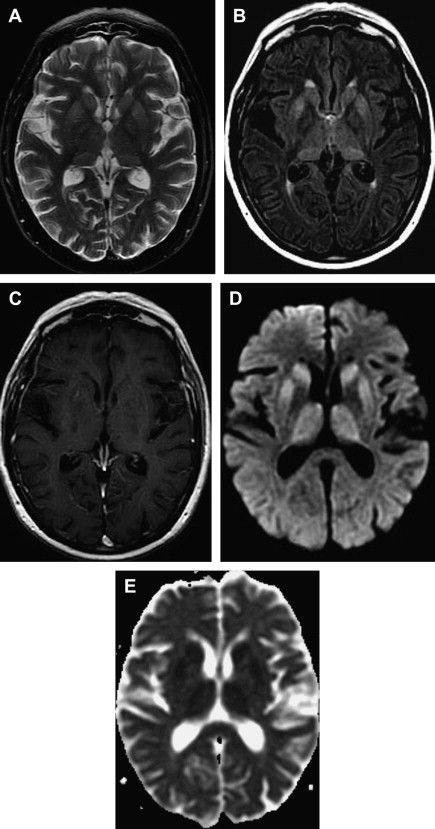
MR imaging has been proved to be an important diagnostic tool in the diagnosis and characterization of human prion disease. Typical MR imaging findings in sCJD are hyperintensity on T2-weighted and FLAIR images in gray matter structures, such as the cortex and basal ganglia. Increased signal on diffusion-weighted imaging (DWI) with restricted diffusion on the apparent diffusion coefficient (ADC) in the basal ganglia and cerebral cortex is also a common feature. These findings are more frequently seen in the head of caudate nuclei, the putamen, the thalamus, the striatum, and the cortical gray matter in sCJD ( Figs. 7 and 8 ) .
Related posts:
 Imaging of Topographic Viral CNS Infections
Imaging of Topographic Viral CNS Infections
 Neuroimaging of Viral Infections in Infants and Young Children
Neuroimaging of Viral Infections in Infants and Young Children
 Imaging of Nonspecific (Nonherpetic) Acute Viral Infections
Imaging of Nonspecific (Nonherpetic) Acute Viral Infections
 Central Nervous System Infections of Herpesvirus Family
Central Nervous System Infections of Herpesvirus Family
 Prion Infections of the Brain
Prion Infections of the Brain
 Prion Protein Disease and Neuropathology of Prion Disease
Prion Protein Disease and Neuropathology of Prion Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


