KEY POINTS
Many congenital anomalies of the brain do not derive from abnormal embryogenesis but are the consequence of destructive processes that may occur any time in gestation, particularly in the third trimester.
Most of these destructive processes are the consequence of vascular accidents, hemorrhage, or occlusion. The etiology is often unknown, but they may derive from a variety of obstetric complications, such as placental insufficiency, coagulation disorders, drug consumption, and transplacental infections.
Disruptive lesions of the fetal brain are clinically important because they may have severe consequences, but they frequently escape early detection.
Intracranial hemorrhage is probably the most common and therefore the best known of all intrauterine disruptions of the fetal brain. The hemorrhage occurs usually into the lateral ventricles, and the sonographic pictures change with time. An echogenic collection is first seen, and in the following days it develops into a complex mass frequently complicated by severe ventriculomegaly.
Prenatal stroke is considered the most important determinant of cystic destruction of the cortex that, depending on the time of occurrence and the severity, may result in a spectrum of conditions, including porencephaly (single or multiple cysts replacing brain parenchyma), schizencephaly (a gray matter–lined cleft in the cerebral mantle connecting the cavity of lateral ventricles to the subarachnoid space), and hydranencephaly (complete destruction of the cerebral hemispheres).
Cerebellar lesions are discussed separately even though they also deal with intracranial hemorrhage. However, they deserve more focused attention.
Intrauterine insults may lead to brain ischemia (stroke), which is a major contributor to the sonographic brain findings that we will discuss in this chapter. Prenatal stroke can be the result of an arterial ischemic event, a venous thrombosis, or hemorrhage. The end-stage lesion is a cavity in the brain tissue of variable size and location.1 The location of the cavity is predictable and stable depending on the vessel that was affected.2 For example, stroke affecting the middle cerebral artery (MCA) will result in porencephaly and that affecting both internal carotid arteries (ICAs) in hydranencephaly. There are several factors determining the propensity of the immature brain to undergo dissolution and eventually cavitation: (1) the high water content of the unmyelinated brain, (2) the relative paucity of myelinated fibers, and (3) deficient glial response. The first two factors result in dissolution of the brain, and the latter is responsible for the cavitation.
Porencephaly is a collective term for a variety of cystic lesions of the brain. Some of these cavities communicate with the ventricular system, the subarachnoid space, or both. These defects have many similarities in etiopathogenesis with schizencephaly and hydranencephaly.1 It is the outcome of an insult such as ischemic stroke, infection, hemorrhage, or trauma occurring between the second trimester of pregnancy and the early postnatal period. This insult results in focal or multifocal areas of brain necroses, which subsequently undergo dissolution and cavity formation.1,2,3
Perencephaly, porencephalia; schizencephaly, porencephalic cyst.
Perinatal arterial ischemic stroke (PAIS) is estimated to occur in 1 in 2500 to 1 in 5000 term neonates. The perinatal period spans from the 28th postmenstrual week of gestation to a week after delivery; it is during this period that stroke is more likely to occur when compared with any other time during childhood. Porencephaly is the end result of a PAIS, and the neonatal outcome is significant neurologic morbidities, such as hemiplegic cerebral palsy (CP).4,5
Porencephaly can be the end-stage result of an ischemic stroke following either an arterial or venous infarction or an intraparenchymal hemorrhage. Areas affected by the infarct undergo tissue necrosis and eventually resorption, leaving behind a cavity in the brain or porencephalic cyst. In “simple” porencephaly, the end result of a venous medullary infarction, typically there is a single cavity along the frontal, parietal, or temporal horns that communicates with the ipsilateral ventricle. Often this ventricle is dilated.1 In preterm neonates, the parenchymal lesions associated with germinal matrix and intraventricular hemorrhages are the result of venous infarct (see below). Eventually these infarcts undergo cystic degeneration; small lesions are seen as periventricular leukomalacia, and large lesions that connect with the ventricles as porencephalic cysts. In most of these cases, the cortical mantle is spared, and the cysts take the shape of the area of the infarct.1
In the arterial type or clastic porencephaly, there is occlusion of an artery often on the left side of the brain; typically, a cavity is seen along the path of the MCA, which is the vessel most commonly involved. However, any arteries, such as the anterior cerebral, posterior cerebral, or anterior choroidal artery, can be affected.1 As eluded above, arterial ischemic stroke occurs in the left hemisphere in ~55% of cases; bilaterally, in ~6%.6 Benders et al6 theorize that this may be the result of the hemodynamic differences from the patent ductus arteriosus or right-to-left intracardiac shunt involving the more direct route of the left common carotid artery.
Thrombophilias, specifically factor V Leiden and antiphospholipid antibodies, may play an important role in the pathogenesis of perinatal stroke; however, at present their role is not completely understood.7
The etiology of ischemic perinatal stroke that eventually may result in porencephaly has not been clearly elucidated. However, there are multiple potential risk factors both maternal and fetal/neonatal that have been associated with this condition (Table 10–1). In addition, thrombotic events on the fetal side of the placenta may potentially result in a thrombotic event due to the patency of the foramen ovale and to the right-to-left direction of the blood flow in the fetal system.5 It has been proposed that in symptomatic cases of ischemic perinatal stroke, a workup similar to that performed on neonates should be done (Table 10–2).5
Maternal factors/conditions Thrombotic disorders (see Table 10–2) Infertility and infertility treatment Preeclampsia Prolonged rupture of membrane (>24 h) Chorioamnionitis Maternal autoimmune conditions and autoantibodies (platelet alloantigen-1) Antiphospholipid syndrome |
Fetal/neonatal disorders Mutations in procollagen IVa1 Inherited thrombophilia Twin-to-twin transfusion syndrome Fetal/neonatal polycythemia Congenital heart disease Neonatal hypoglycemia (in preterm infants) Persistent fetal circulation and extracorporeal membrane oxygenation therapy Intrauterine growth restriction Fetal/neonatal infections and meningitis |
Nonspecific factors Ethnicity and race (higher incidence in black infants compared with non-Hispanic white infants) Infant gender (higher incidence in boys) |
| Plasma/Protein Based | DNA Based |
|---|---|
| Activated protein C resistance | Factor V G1691A |
| Protein C activity/antigen | Prothrombin G20210A |
| Free and total protein S antigen | |
| Antithrombin activity/antigen | |
| Lipoprotein (a) | |
| Fasting homocysteine | |
| Lupus anticoagulant/antiphospholipid antibodies | |
| Fibrinogen (Clauss) | |
| Plasminogen | |
| Factor VIIIC |
Porencephaly has also been described as the result of several other types of intrauterine exposures or insults. A more recent report8 documents maternal carbon monoxide poisoning at 22 postmenstrual weeks resulting in the prenatal diagnosis of porencephaly. Another case report9 described a patient who was treated with warfarin for a prosthetic heart valve and at 22 postmenstrual weeks suddenly had a surge on the prothrombin International Normalized Ratio (INR) to double its previous values. At 26 postmenstrual weeks, an ultrasound (US) demonstrated a large intracranial echogenic lesion suggestive of a subdural hematoma, and at birth the magnetic resonance imaging (MRI) revealed porencephalic cyst and mild ipsilateral ventriculomegaly. A case report10 documents a prenatally detected case of porencephaly at 28 weeks following inadvertent penetration of the fetal skull during an amniocentesis unguided by continuous US at 16 weeks. Initially, the head US was normal, but at 28 weeks a left-sided ventriculomegaly and an anechoic mass in the area of the lateral ventricle were noted and confirmed at birth by computed tomography (CT) and MRI studies. Other reported events that have resulted in porencephaly are chorionic villus sampling, cocaine, vitamin A, and valproate use.8 Porencephaly and other cystic brain lesions are seen frequently in monozygotic twins.1 Familial cases of porencephaly have also been described.11,12,13,14
With the exception of ventriculomegaly, there are no typical associated anomalies in cases of porencephaly, but in the arterial type or classic porencephaly, areas of polymicrogyria may border the cysts.1
At present there are no data regarding the risk of recurrence of a perinatal stroke, but recurrent stroke is rare among infants who have suffered a perinatal stroke.15 The risk of recurrence of stroke in children who have suffered a perinatal stroke ranges from 3% to 30%.15,16
Among fetuses/neonates who have suffered a perinatal arterial stroke and their mothers, there is a high rate of thrombophilias when compared with the general population. Simchen et al7 found that 64% of infants with perinatal arterial stroke had at least one thrombophilic marker, and among the mothers, 68% were carriers of a thrombophilia. In their study, factor V Leiden mutation, protein C deficiency, and the presence of antiphospholipid antibodies were significant factors for perinatal stroke. The authors recommended that any child that has suffered a perinatal arterial stroke have both parents tested for thrombophilias.
A mutation in collagen IV A1 (COL4A1) gene has been reported in a few families with an autosomal dominant form of porencephaly in which the porencephalic cyst is the result of a perinatal hemorrhage.17 In cases of familial autosomal dominant porencephaly (OMIM 175780), the inheritance is similar to any dominant disease in which each baby has a 50% chance of receiving the affected mutation.
The sonographic appearance of porencephaly is that of a cystic lesion that communicates with the lateral ventricle (Figure 10–1). The term porencephaly is derived from the Latin word porus, meaning communication between the ventricular and extracerebral space. The ipsilateral ventricle is dilated. The porencephalic cyst never causes a mass effect and is typically located along the distribution of the middle cerebral artery or other arteries (see above). This helps differentiate it from arachnoid and interhemispheric cysts.18

Porencephaly may be missed by antenatal sonography, particularly in early gestation, because of two reasons. First, it is usually a unilateral lesion; this makes it difficult to demonstrate when it occurs in the hemisphere proximal to the transducer, being usually obscured by sound reverberation and artifacts.19 Second, it usually occurs only in late gestation.1
The differential diagnosis of porencephaly includes all cystic brain lesions (see Chapter 9), but the most important differential diagnosis is the arachnoid cysts and unilateral schizencephaly. Arachnoid cysts are collections of cerebrospinal fluid (CSF). They are usually benign, congenital, space-occupying lesions; unlike porencephaly they do not communicate with the ventricles. The cyst wall is lined with collagen and cells of the arachnoid matter. In the arachnoid cyst, the CSF is located within the layers of the arachnoid membrane, which may or may not communicate with the subarachnoid space. In unilateral schizencephaly, the cyst communicates with the subarachnoid space, and the cavity is lined by gray matter; this is easily seen during a fetal MRI. Cystic neoplasms are rare, and these usually have mass effects with both solid and cyst components.
Porencephaly is associated with significant morbidity and mortality. Ischemic perinatal stroke resulting in porencephaly is the leading cause of cerebral palsy (CP), and congenital hemiplegia is the most common type of CP.5,20 Hemiparesis and motor deficits are seen in >80% of the presumed perinatal ischemic stroke.5,20 In addition, 50% to 75% of survivors of perinatal ischemic stroke will have neurologic deficits or epilepsy. Moreover, ~20% to 60% of survivors will have deficits in language, vision, cognition, and behavior.5 Unfortunately, there are no clearcut figures. Most prenatally diagnosed cases tend to have a poor outcome.21,22,23
Advances in neuroimaging have facilitated outcome prediction in cases of perinatal stroke, as described by Kirton and deVeber.20 Outcome prediction provides important information for the family and allows patients to be entered into appropriate clinical trials. For example, lesion size and location are somewhat correlated with clinical outcomes. Poor motor outcomes can be predicted by infarction lesions of the MCA, periventricular venous lesions, or basal ganglia involvement, whereas isolated subcortical lesions carry a low risk of language, cognitive deficits, or epilepsy.
In cases where porencephaly is diagnosed early (earlier than 24 weeks’ gestation), termination of pregnancy should be offered to the patient; however, in the vast majority of cases, porencephaly will be diagnosed only during the third trimester; in these cases, management of the pregnancy with porencephaly should include a thrombophilia workup ideally of both parents, fetal MRI to further evaluate the fetal brain, and consultations with a geneticist, neonatologist, pediatric neurologist, and neurosurgeon. Given the fact that porencephaly is a relatively rare condition, there are no standard recommendations at this time regarding the best route of delivery. It is our opinion that, given the significant morbidities associated with porencephaly, cesarean section should be performed for routine obstetric indications.
Schizencephaly is defined as a transcerebral, full-thickness, gray matter–lined clefts or defects extending from the lateral ventricles to the pial surface of the brain. The clefts of schizencephaly can be unilateral or bilateral and open or closed. In closed-lip schizencephaly (or type I), the lips of the cleft touch or are fused with each other (Figure 10–2); in contrast, in open-lip schizencephaly (or type II), the walls of the clefts are widely separated, and the space is filled with CSF, which is contiguous from the lateral ventricles to the subarachnoid space (Figure 10–2). Type II is frequently seen with hydrocephaly. Although schizencephaly can occur anywhere in the cerebral hemispheres, it is more commonly seen in the perisylvian area.24

True porencephaly; early fetal porencephaly.
Schizencephaly is a rare brain abnormality that was first described by Yakovlev and Wadsworth in 1946.25 A recent publication of the birth prevalence of schizencephaly in south-eastern Hungary found it to be present in 0.54 per 10,000 live births;26 this is in contrast with a study from the California Birth Defects Monitoring Program that found a prevalence of 1.54 per 100,000 (0.15 per 10,000).27 A major difference between the studies was that the California study included patients up to 1 year of age, whereas in the Hungarian study, the average age at confirmation was 28.7 months; as a result, the California study may have underreported mild cases.26 In the majority of patients, schizencephaly is sporadic, but familial schizencephaly has been reported.24
There are two main theories regarding the pathogenesis of schizencephaly. The first is that of a failure of induction of neuronal migration; the second is that of vascular disruption and hypoxia-ischemia at critical points during the neuronal development (acquired). These processes usually occur before the 24th week of the pregnancy.26,28
The etiology of schizencephaly is heterogeneous and is not clear at present. Etiologies reported in the literature include viral teratogenicity as the result of in utero exposure to cytomegalovirus (CMV), warfarin exposure, alcohol abuse, cocaine use, trauma during the first and second trimesters, syndromic associations, association with the EMX2 gene, maternal and/or infant thrombophilia, and alloimmune thrombocytopenia (hemorrhage).27
Curry et al27 reported on 63 cases of schizencephaly from the California Birth Defects Monitoring Program; 43 cases had schizencephaly and central nervous system (CNS) anomalies only. The more common CNS anomalies present were hypoplastic or absent corpus callosum, absence of the septum pellucidum, hydrocephaly, gyral malformations (including heterotopias and polymicrogyria), and optic nerve hypoplasia. Rarely seen CNS anomalies included fusion of the thalami, accompanying porencephaly, arachnoid cyst, and cerebellar malformations. There were 20 cases that in addition to the associated CNS anomalies had non-CNS anomalies, such as amniotic band disruptive sequence, arthrogryposis, death of a monozygotic twin, septo-optic dysplasia, gastroschisis, cleft lip and/or palate, Aicardi syndrome, meningocele, 8p+, VATER association (vertebral defects, imperforate anus, tracheoesophageal fistula, radial and renal dysplasia), craniosynostosis, microphthalmia, cataracts, and hydronephrosis. In a more recent study by Szabo et al,26 ~50% of the cases of schizencephaly had associated agenesis of the septum pellucidum; however, none of the patients had optic nerve hypoplasia or endocrinological abnormalities, which are typically seen in septo-optic dysplasia. In ~20% there was polymicrogyria contralateral to the cleft and agenesis or dysgenesis of the corpus callosum; there was also one case each of crossed cerebellar diaschisis and intracerebral calcification in the absence of any intrauterine infections.
The risk of recurrence is uncertain at this time, as most cases are sporadic; familial cases of schizencephaly have been described. Some patients with schizencephaly have been found to have mutations in the EMX2 gene (OMIM 269160).
The sonographic diagnosis is that of bilateral or unilateral wedge like defects or clefts in the cerebral cortex extending from the lateral ventricles to the subarachnoid space (Figure 10–3). The cavum septi pellucidi and the corpus callosum may be absent; there may be optic nerve hypoplasia. The thalami typically are not fused. The ventricles may be dilated. The circle of Willis is normal.29



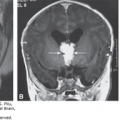
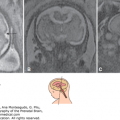
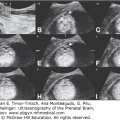
Fetal MRI is helpful in identifying the gray matter. In open-lip schizencephaly, the gray matter is seen lining the walls of the clefts. In addition, it can help in identifying areas of polymicrogyria and heterotopias, which are common in cases of schizencephaly.30 Oh et al30 described the following postnatal MRI findings in schizencephaly: (1) a defect that extends from the pial surface to the ventricle; (2) the walls of the defect are lined with gray matter; (3) the ventricle may be tented, thus pointing to the defect; (4) absent cavum septi pellucidi in as many as 75% of cases of schizencephaly; (5) the corpus callosum is focally thinned or may be absent; (6) polymicrogyria and heterotopias are common; and (7) a roofing membrane covering the defect is infrequent (Figures 10–4, 10–5, and 10–6). Three-dimensional (3D) US helps to better define the defect, and pictures are comparable to MRI (Figures 10–7, 10–8, and 10–9).



Figure 10–6.
Comparative US (upper row) and MRI (lower row) of a unilateral, right-sided, open-lip schizencephaly. Sagittal, coronal, and axial planes, respectively (same case as in Figures 10–7 and 10–8).

Figure 10–7.
3D US images of a unilateral, right-sided, open-lip schizencephaly. The arrows point to the cleft (same case as in Figures 10–6 and 10–8).

Figure 10–8.
3D inversion rendering of the fluid in a fetal brain demonstrating the fluid connected between the lateral ventricles and the subarachnoid space through the cleft (same case as in Figures 10–6 and 10–7).


Schizencephaly occurs very early in gestation. It is therefore likely that the cerebral clefts associated with the type II variety can be recognized by the midtrimester. Absence of the corpus callosum and ventriculomegaly are also frequently present, and this would facilitate the diagnosis. An exception could be represented by cases with unilateral clefts involving the cerebral hemisphere proximal to the transducer that is commonly not seen during standard examinations.19 Most cases thus far have been recognized only in late gestation.
The main differential diagnoses include holoprosencephaly, hydrocephaly, hydranencephaly, porencephaly, and arachnoid cysts (see Chapter 9). In holoprosencephaly, there is absence of the midline structures, fused thalami, and facial abnormalities (alobar and semilobar type); however, in schizencephaly, although the cavum septi pellucidi and corpus callosum may be absent, there are large cortical abnormalities, and typically the thalami are not fused. In hydrocephaly, the dilated lateral ventricles do not communicate with the subarachnoid space. In cases of hydranencephaly, the cerebral hemispheres may be completely or almost completely absent, and CSF fills the space. In porencephaly, although it may have a similar appearance, the porencephalic cavities are not lined by gray matter; this can be diagnosed by using MRI. Arachnoid cysts are not symmetrical and do not communicate with the lateral ventricles.
The clinical symptoms correlate with the degree and severity of the cleft coupled with the severity of the cortical abnormalities.24 Clinically, schizencephaly is typically characterized by a triad of abnormalities of neuronal migration, namely, motor disorder, such as hemi- or tetraparesis; intellectual impairment; and seizures.31 Epilepsy is usually seen in children with unilateral closed schizencephaly, and microcephaly, spastic quadriplegia, and mental retardation in children with bilateral open clefts.1 In a recent study by Szabo et al,26 of 10 children with schizencephaly, 7 exhibited unilateral schizencephaly, and 3 had bilateral. Of the seven children with unilateral schizencephaly, six had contralateral spastic hemiplegia, and one had spastic tetraplegia; five of the seven had delayed development and intellectual disability, and two of the seven had seizures. Of the three children with bilateral schizencephaly, two had spastic tetraplegia, and the other had generalized hypotonia; all three had delayed developmental and intellectual disability, and one had seizures.
Schizencephaly is a rare heterogeneous disease; if diagnosed early (earlier than 24 postmenstrual weeks), termination of pregnancy should be offered to the patient; however, in the vast majority of cases, schizencephaly, like porencephaly, will be diagnosed only during the third trimester of pregnancy. In these cases, management should include fetal MRI to further evaluate the fetal brain, a thrombophilia workup (for both parents),32 and consultations with a geneticist, neonatologist, pediatric neurologist, and neurosurgeon. Given the fact that schizencephaly is a rare condition, there are no standard recommendations at this time regarding the best route of delivery.
Hydranencephaly is a rare brain malformation, the result of an encephaloclastic (destructive) process in which both cerebral hemispheres may be completely or almost completely absent, and CSF fills the space; essentially, the cerebral hemispheres have been reduced to a CSF-filled sac. The falx may be incomplete or absent, and the head size may be normal. The cerebellum and brainstem are normal but subject to intracranial pressure and are therefore deformed.
Hydrocephalic anencephaly, hydroencephalodysplasia, cystencephaly hydromercencephaly, encephaloclastic proliferative vasculopathy (EPV), hydranencephaly, Fowler syndrome
Hydranencephaly occurs rarely, with a reported prevalence of 1 to 2 per 10,000 births. Most cases are sporadic; however, familial types of hydranencephaly have been described, such as (1) proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (MIM 225790), also known as Fowler syndrome, which is a recessive condition (gene map locus14q24.3); another feature of the syndrome is fetal akinesia deformation sequence, which can be seen from the late first to early second trimester;33 (2) micro hydranencephaly (MIM 605013), in which there is microcephaly and hydranencephaly (gene map locus16p13.3-p12.1); and (3) hydranencephaly with renal aplasia-dysplasia (MIM 236500).
Hydranencephaly is the result of an ischemic stroke of both internal carotid arteries (ICAs) occurring as early as the 11th postmenstrual week.1,34 The areas affected by the infarct undergo tissue necrosis and eventually resorption, leaving behind a cavity in the brain. In hydranencephaly, the previously normal cerebral hemispheres are replaced by a thin-walled, fluid-filled cyst. The aqueduct is usually atretic, and increased fluid pressure causes the cyst (and the head) to enlarge as the result of hydrocephaly. There is variable preservation of the inferior frontal, temporal, and occipital lobes and of the basal ganglia and diencephalon. The brainstem and cerebellum are usually preserved. If a unilateral internal carotid stroke occurs, the result is hemihydranencephaly; usually the left side is affected. In contrast, with children with hydranencephaly, those with hemihydranencephaly have a relatively good outcome.1
Hydranencephaly can be the result of a developmental or an encephaloclastic process secondary to an insult, such as vascular occlusion of a vessel of the anterior circulation (eg, the vein of Galen or ICAs), resulting in ischemia; intracranial hemorrhage, as in cases of coagulation disorder or thrombocytopenia; intrauterine infection (eg, herpes, CMV, or toxoplasmosis); and maternal-fetal hypotension, such as in the death of a monochorionic co-twin, maternal trauma, or abruption.1,35,36
Hydranencephaly can be associated with other brain anomalies, such as hydrocephaly, polymicrogyria, and microcephaly. When non-CNS abnormalities, such as fetal akinesia deformation sequence, are seen, the diagnosis of Fowler syndrome should be entertained. Renal abnormalities, such as hypoplastic kidneys, can also be observed with hydranencephaly as part of extremely rare syndromes.
The risk of recurrence is uncertain, as most cases are sporadic. However, when hydranencephaly is part of a syndrome, such as Fowler (proliferative vasculopathy and hydranencephaly-hydrocephaly), it has a recessive inheritance.
The sonographic findings of hydranencephaly include a normocephalic or macrocephalic fetus with a large fluid-filled intracranial cavity (Figures 10–10 and 10–11). Several reports on the in utero evolution of hydranencephaly resulting from an intracranial hemorrhage are present in the literature.37,38 The usual presentation is the finding of a bright, homogeneous, hyperechoic mass most consistent with an intracranial hemorrhage. As the cases are followed prospectively with serial sonography, the echo-dense mass becomes more sonolucent with the development of the hydranencephaly. As a rule, no cerebral cortex is present, but there is partial preservation of portions of the occipital lobe. The falx cerebri is usually intact. The midbrain and basal ganglia are variably preserved. The brainstem and cerebellum are usually intact.38 Polyhydramnios is usually present.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


