Lesions of Uncertain Differentiation
The World Health Organization (WHO) previously designated the lesions detailed in this chapter as tumors of uncertain histogenesis, but now refers to them as tumors of uncertain differentiation. The change in nomenclature, although seemingly small, reflects a conceptual change in the origin of soft tissue tumors. Rather than arising from their normal cellular counterparts, soft tissue tumors differentiate by patterns determined by their genetics (1). Tumors classified as “uncertain differentiation” are those in which there is no clear line of cellular differentiation or normal cellular counterpart that the tumor is recapitulating (1).
Some of the lesions classified by the WHO to be of uncertain differentiation are not included in this chapter, but covered instead in other sections of the text. Rather than strictly follow the WHO classification, we have taken liberties, including some of those lesions identified as tumors of uncertain differentiation, with other groups, when such is useful for purposes of differential diagnosis.
BENIGN LESIONS
Benign tumors of uncertain histogenesis covered in this chapter include myxoma, deep aggressive angiomyxoma, ectopic hamartomatous thymoma, and mixed tumor/myoepithelioma/parachordoma.
Myxoma
Several lesions are characterized by an abundant myxoid matrix and a paucity of spindle-shaped stromal cells. Virchow first introduced the term myxoma in 1863, describing a benign tumor which histologically resembles the mucinous substance of the umbilical cord (2, 3). It was Stout in 1948, however, who established myxoma as a distinctive lesion, in a comprehensive report of 49 cases from the Laboratory of Surgical Pathology of Columbia University and 93 additional cases from the literature (exclusive of those within the heart) (4). Although only four (3%) of the lesions in Stout’s original report were intramuscular, the intramuscular myxoma is the best known of these myxoid lesions to radiologists and the one most likely to mimic a myxoid musculoskeletal sarcoma (5, 6, 7, 8). A subsequent report of almost 200 myxomas of various sites from the Armed Forces Institute of Pathology (AFIP), Washington, DC, identified 34 (17%) as intramuscular in origin (9). The reported annual incidence is one case per million people (5, 6, 7, 8). Other locations of noncardiac myxoma, in decreasing order of frequency, include subcutaneous and aponeurotic tissue (22%), bone (18%), genitourinary tract (16%), and skin (15%) (9). The more recent study of 45 cases by Murphey et al. likely reflects the distribution of lesions seen by radiologists more accurately, showing that 82% of musculoskeletal myxomas were intramuscular, 9% intermuscular, and 9% subcutaneous; three of these cases were also juxta-articular (10).
Intramuscular Myxoma
In a review of 34 cases in 1965, Enzinger (9) defined intramuscular myxoma as a distinctive benign lesion, speculating that the myxoma cell is an altered fibroblast that produces excess mucopolysaccharide and is incapable of assembling mature collagen. It is a lesion usually seen in adults, with a peak presentation between the fifth and seventh decades (5, 6, 7, 8). Less commonly, it is seen in young adults and rarely in children (11). In most series, women are affected more commonly than men (2:1 ratio) (9, 11, 12, 13). A history of trauma is elicited in fewer than 25% of patients (5, 6, 7, 8).
Patients typically present with a painless, palpable mass and less than 25% complain of pain or tenderness (9). The rate of tumor growth is variable, and there may be long intervals with no apparent clinical enlargement (5). Intramuscular myxomas are usually solitary, although multiple tumors are reported. Multiple myxomas are almost always associated with fibrous dysplasia of bone. This association is referred to as Mazabraud syndrome and is discussed in greater detail later in this chapter. Intramuscular myxoma is most common in the thigh, with more than 50% of
lesions occurring in this location (5, 9, 12). Less common locations, in order of decreasing frequency, include the shoulder, buttocks, and upper arm (5). Rare case may be seen in the neck and paraspinal muscles (14).
lesions occurring in this location (5, 9, 12). Less common locations, in order of decreasing frequency, include the shoulder, buttocks, and upper arm (5). Rare case may be seen in the neck and paraspinal muscles (14).
KEY CONCEPTS
Lesions are most commonly seen in adults (fifth to seventh decades); rarely in children.
Patients typically present with a painless, palpable mass; less than 25% complain of pain or tenderness.
Simple excision is usually curative, without recurrence.
Radiographs are typically normal or may show a nonspecific mass.
Bone scintigraphy usually shows only mild or no uptake of radionuclide.
Ultrasound will show a well-defined hypoechoic to near anechoic mass, with some internal echoes and increased through transmission; small anechoic cystic areas are seen in the majority of cases.
CT typically reveals a well-defined, homogeneous, soft tissue mass with attenuation greater than that of water and less than that of surrounding normal muscle.
MR will show a homogeneous or mildly heterogeneous fluid-like mass.
Lesions are well-defined in 60% to 80% of cases.
A small rim of fat representing atrophied surrounding muscle is identified in 65% to 89% of cases.
This fat rim is usually most prominent at the superior and inferior extent of the lesion.
Surrounding high signal, caused by leakage of the myxomatous tissue into surrounding muscle, is usually seen.
At gross pathologic inspection, myxomas are composed of a gelatinous material and are usually 5 to 10 cm in size (5, 6, 7, 8). Microscopically, they are hypocellular, consisting of scattered, bland, spindle and stellate cells within a loose, highly myxoid, avascular stroma (15, 16). Associated cystic changes may be seen (15). Although intramuscular myxoma appears well-circumscribed both grossly and radiographically, microscopically it is unencapsulated, with infiltrating margins, blending imperceptibly with the adjacent muscle and fascia (5, 15). The myxoid tissue often extends into the surrounding muscle, because lesions lack a complete pseudocapsule and may result in muscle atrophy about the rim of the tumor (Fig. 11.1) (17). The bland appearance of the cells and the highly myxoid stroma often suggest the diagnosis of myxoid liposarcoma to the pathologist. However, the absence of a plexiform vasculature and lipoblasts is in sharp contrast to the findings in myxoid liposarcoma (15). In fact, the absence of an elaborate vasculature has suggested that intramuscular myxoma may not be a true neoplasm but rather a localized accumulation of reactive fibroblasts and acid mucopolysaccharides (15). However, Nielsen et al. identified more hypercellular regions (10% to 80% of the lesion volume) in 76% of cases with relatively increased vascularity (18). Mutations in the GNAS1 gene are described with myxomas, similar to genetic aberrations in fibrous dysplasia (6).
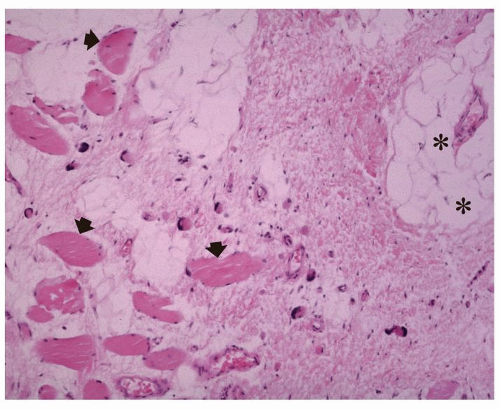 FIGURE 11.1 Intramuscular myxoma: Histology. The lesion is characterized by a paucity of cells and an abundance of mucoid material. Fluid-filled cystic spaces (asterisks) are seen as is atrophy of the skeletal muscle (arrows). Note increased fat cells associated with the atrophic muscle at the periphery of the mass. |
Despite the occasional large size of these tumors, simple excision, even if not complete, is usually curative (12, 19, 20, 21, 22, 23). Nielson et al. reported no recurrences in 32 cases (18), whereas Ireland et al. reported only two local recurrences in 39 cases (5%) (11). Because of the limited growth potential, we believe observation of these lesions following biopsy may be appropriate in some patients. Metastases are not reported (21).
Radiographs may be normal (55%) or may reveal a nonspecific soft tissue mass (45%) (10). However, Ireland et al. (11) described a case of intramuscular myxoma in which the tumor was less dense than the surrounding muscle, having the radiographic appearance of a lipoma. Internal calcification is uncommonly reported (20, 24). Miettinen et al. (13) reported the radiographic findings in 16 patients and found minor bone abnormalities in 14, including cortical thickening, exostoses, subchondral cysts, and supernumerary bones. We believe associated osseous findings are rare with myxoma. Bone scintigraphy usually shows only mild or no uptake of radionuclide scintigraphy. Arteriography may reveal an avascular or hypovascular lesion.
Ultrasonography is nonspecific, showing a welldefined hypoechoic to near anechoic mass, with some internal echoes and increased through transmission (15, 25, 26). Small, anechoic, cystic areas may be seen in the majority of cases (83%) (Fig. 11.1) (10). Color Doppler sonography demonstrates lesions to be avascular or hypovascular (10).
Computed tomography (CT) typically reveals a well-defined, homogeneous, soft tissue mass with attenuation greater than that of water and less than that of surrounding normal muscle (Figs. 11.2 and 11.3) (15, 20, 24, 27, 28). A lesion with a CT attenuation coefficient of −53 Hounsfield units, a level approaching that of fat, has also been reported (15). In this case, the low attenuation may be the result of volume averaging of the fat atrophy surrounding the lesion, as can be seen in 25% of cases, rather than from the lesion itself (Fig. 11.4) (10). Mild enhancement
following contrast, either diffuse, or septal and peripheral, is seen in 50% of cases (10).
following contrast, either diffuse, or septal and peripheral, is seen in 50% of cases (10).
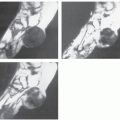
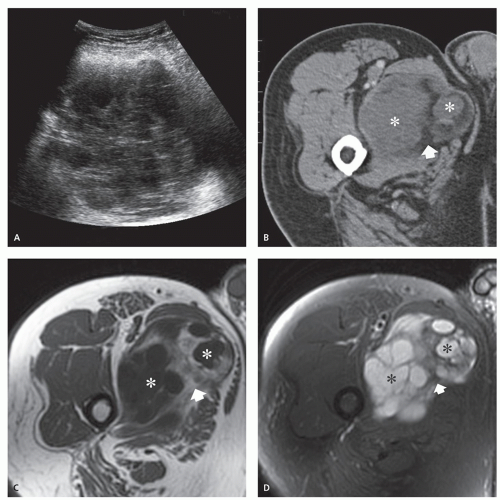 FIGURE 11.2 Intramuscular myxoma: Imaging features in a 56-year-old man with a painless, slowly enlarging mass of the adductor muscles. A: Ultrasonography shows a well-defined, hypoechoic mass, with some internal echoes and increased through transmission. B: CT reveals a well-defined, relatively homogeneous, lobulated, soft tissue mass with attenuation greater than that of water and less than that of surrounding normal muscle (asterisks). Note rind of fat within the lesion, representing atrophied muscle extending between lobules of the tumor (arrow). Axial T1-weighted (TR/TE; 450/12) (C) spin-echo and short-tau inversion recovery (STIR) (TR/TE; 2,500/65) (D) MR images show a lobulated mass (asterisks) with fluid-like signal intensity, with intermixed fat (arrows). (continued) |
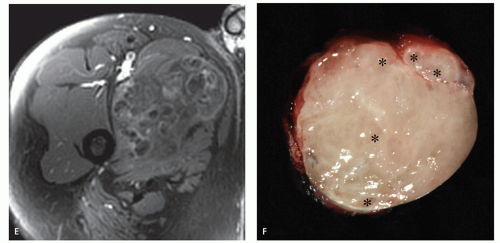 FIGURE 11.2 (Continued) E: Corresponding enhanced fat-suppressed T1-weighted spin-echo (TR/TE; 660/12) image shows areas with moderate diffuse enhancement as well as those with thin peripheral enhancement, representing the more cellular and cyst-like areas of the tumor respectively. F: Bisected gross specimen shows the myxoma with multiple cystic area (asterisks) corresponding to the imaging findings. |
Multiple studies (10, 19, 22, 29, 30, 31, 32, 33, 34) have described the MR imaging appearance of myxomas. Myxomas show low (81% to 100%) to intermediate (0% to 19%) signal intensity on T1-weighted MR images (10, 32, 33, 34). On T2-weighted MR images, all myxomas show high signal intensity (10, 32, 33, 34) (Figs. 11.5, 11.6, 11.7). Lesions are typically homogeneous or mildly heterogeneous, and are well-defined in 60% to 80% of cases (10, 32, 33, 34). A small rim of fat representing atrophied surrounding muscle, as is seen pathologically, can be identified in 65% to 89% of cases (10, 32, 33, 34) (Figs. 11.2 and 11.5). This fat rim is usually most prominent at the superior and inferior extent of the lesion. Surrounding high signal caused by leakage of the myxomatous tissue into surrounding muscle may be seen in 79% to 100% of myxomas (10, 32, 33, 34, 35) (Fig. 11.7). Following contrast administration, lesions will show mild (76%) to moderate (24%) enhancement, in either a diffuse (57%) or thick peripheral and septal (43%) pattern (Fig. 11.5). Cystic areas can be seen in 52% of myxomas by MR imaging (10). We believe the MR appearance of an intramuscular mass with low signal intensity on T1 weighting, high signal intensity on T2 weighting, and a peripheral rim of fat and surrounding edema is nearly pathognomonic of myxoma. These features are not apparent in other intramuscular masses (36, 37).
Myxoma and Fibrous Dysplasia (Mazabraud Syndrome)
The association of fibrous dysplasia with soft tissue myxoma is well-established, although it is uncommon. First described by Henschen in 1926 (38), this association was emphasized by Mazabraud et al. in 1967 (39) and is often referred to as Mazabraud syndrome (40, 41, 42, 43, 44, 45, 46, 47). Endo et al. (48) reported two cases of Mazabraud syndrome in 2007, and reviewed the previously reported 56 cases. This review demonstrated that soft tissue myxoma was
much more common with the polyostotic form of fibrous dysplasia (84%) but was reported with monostotic involvement as well (16%) (16, 48). Ireland et al. (11) reported 58 patients with soft tissue myxoma, 3 of whom had multiple tumors associated with fibrous dysplasia of bone (2 of these 3 patients had polyostotic disease) (11). The myxomas occur in the vicinity of the most severely affected bones and are multiple in approximately twothirds of cases (16, 19, 48). Fibrous dysplasia is typically diagnosed well before the myxomas develop. The association between these entities is suggested to be the result of a common origin for both fibrous dysplasia and myxoma (39), as well as a metabolic anomaly in the initial growth of both bone and soft tissue (49).
much more common with the polyostotic form of fibrous dysplasia (84%) but was reported with monostotic involvement as well (16%) (16, 48). Ireland et al. (11) reported 58 patients with soft tissue myxoma, 3 of whom had multiple tumors associated with fibrous dysplasia of bone (2 of these 3 patients had polyostotic disease) (11). The myxomas occur in the vicinity of the most severely affected bones and are multiple in approximately twothirds of cases (16, 19, 48). Fibrous dysplasia is typically diagnosed well before the myxomas develop. The association between these entities is suggested to be the result of a common origin for both fibrous dysplasia and myxoma (39), as well as a metabolic anomaly in the initial growth of both bone and soft tissue (49).
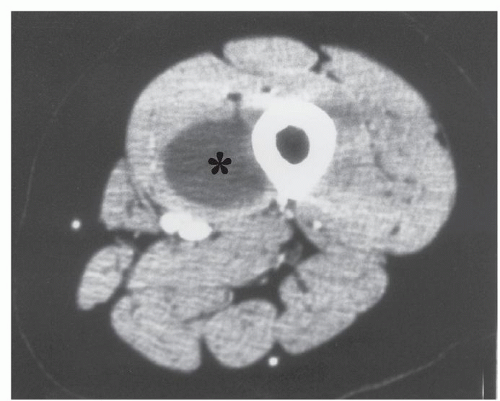 FIGURE 11.3 Intramuscular myxoma: CT appearance in a 58-year-old woman. Axial contrast-enhanced CT scan shows a well-defined mass (asterisk) with prominent low attenuation greater than that of water and less than that of surrounding normal muscle. |
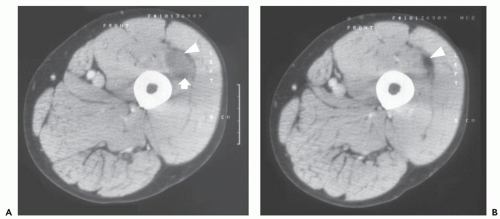 FIGURE 11.4 Intramuscular myxoma: Atypical CT features in a 66-year-old man with a painless thigh mass. A, B: CT at two different levels shows the low attenuation soft tissue mass (arrow) with a small rim of subtle surrounding fat (arrowhead). |
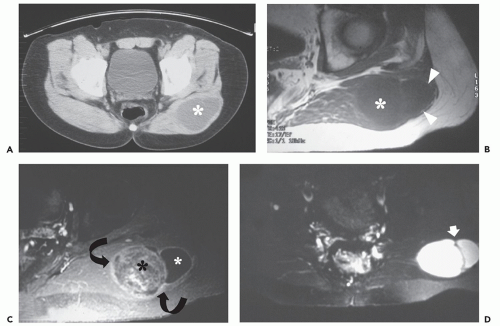 FIGURE 11.5 Intramuscular myxoma. CT and MR imaging features in a 40-year-old man, with a painless, slowly enlarging, gluteal mass. A: CT shows a low attenuation intramuscular mass (asterisk). Axial T1-weighted (TR/TE; 416/17) spin-echo MR images preceding (B) and with fat suppression following (C) intravenous contrast show a low-intensity intramuscular mass (asterisk) with an incomplete rim of fat laterally (arrowheads). The image after contrast shows two distinct areas with moderate diffuse enhancement of the myxoma medially (black asterisk) and thin peripheral enhancement of the cyst laterally (white asterisk). The peripheral enhancement about the entire lesion represents the pseudocapsule (black curved arrows). D: Axial fat-suppressed T2-weighted (TR/TE; 3,850/135) MR image reveals diffuse high signal intensity in both components, separated by a low signal septation (arrow). |
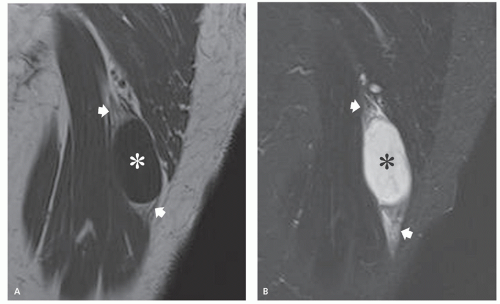 FIGURE 11.6 Intramuscular myxoma. MR imaging features in a 76-year-old woman presenting with a painless, buttocks mass. Sagittal T1-weighted (TR/TE; 500/16) (A) spin-echo and STIR (TR/TE; 2,500/90) (B) MR images of the buttocks show a fluid-like mass (asterisks) in the biceps femoris muscle. Note fat infiltration in the atrophic muscle (arrows), best seen at the ends of the lesion. |
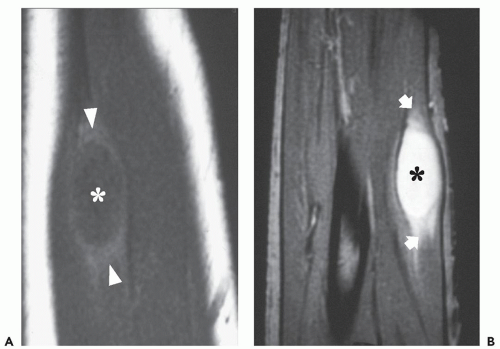 FIGURE 11.7 Intramuscular myxoma. MR imaging features in a 40-year-old woman with a forearm mass. Sagittal T1-weighted (TR/TE; 600/20) (A) and T2-weighted (TR/TE; 2,500/90) (B) MR images of the forearm show a well-defined, cyst-like mass (asterisk). There is a small rim of tissue with signal intensity similar to that of fat on T1-weighted image (arrowheads) and mild surrounding edema on T2 weighting (arrows), and both these features are most prominent at the superior and inferior poles of the lesion. |
KEY CONCEPTS
The rare association of fibrous dysplasia with soft tissue myxoma is known as Mazabraud syndrome.
Mazabraud syndrome is much more common with polyostotic fibrous dysplasia (84%).
The myxomas occur in the vicinity of the most severely affected bones and are typically multiple.
Patients with fibrous dysplasia and myxomas may be at greater risk for malignant transformation than patients having fibrous dysplasia alone.
Malignant transformation in fibrous dysplasia is relatively rare, with a reported prevalence of approximately 0.5%, although two cases of malignant transformation were reported in patients with Mazabraud syndrome (50, 51). This suggests that patients with fibrous dysplasia and myxomas may be at greater risk for malignant transformation than patients having fibrous dysplasia alone (50).
In our experience, all myxomas, regardless of location (intramuscular, subcutaneous, aponeurotic, or juxtaarticular) or associated disease (fibrous dysplasia) have a similar intrinsic imaging appearance (Fig. 11.8).
Subcutaneous and Aponeurotic Myxoma
As with other superficial lesions, these are typically treated on the basis of clinical findings and are rarely imaged radiologically. These lesions arise more frequently in middle-aged males. Sites of involvement include the trunk, lower extremities, and head/neck. Lesions have also been reported in the hands and feet (52). Lesions in the eyelid region are described in association with Carney complex (cutaneous and cardiac myxomas, spotty pigmentation, and endocrine overactivity). Subcutaneous lesions may also be referred to as superficial angiomyxoma or extramuscular myxoma (5, 6, 8, 52, 53). Subcutaneous and aponeurotic myxomas make up approximately 25% (9) of all myxomas. Digital myxomas are also described, although many of these lesions may represent mucoid cysts related to osteoarthritis. Cutaneous myxomas have an increased likelihood of local recurrence (30% to 40%) (5, 6, 7, 8). The pathologic and imaging appearances of these lesions are similar to their intramuscular counterparts (Fig. 11.9).
KEY CONCEPTS
Subcutaneous and aponeurotic myxomas make up approximately 25% of all myxomas, but are rarely imaged.
Sites of involvement include the trunk, lower extremities, and head/neck.
Lesions in the eyelid region are described in association with Carney complex.
Cutaneous myxomas have an increased likelihood of local recurrence (30% to 40%).
The pathologic and imaging appearance of these lesions are similar to their intramuscular counterparts.
The terms superficial angiomyxoma or cutaneous myxoma are usually reserved for those superficial myxoid lesions that occur in the setting of the Carney complex (5). Lesions associated with Carney complex are often multiple and have a propensity for the head and neck, especially the eyelids and external ears. Such lesions are very small, measuring only a few millimeters (53, 54).
Juxta-articular Myxoma
The juxta-articular myxoma, also known as a periarticular myxoma, usually occurs around large joints (particularly the knee). Other locations include the elbow, shoulder, ankle, and hip. These lesions have some histologic features of a myxoma but are frequently associated with prominent cystic change, closely resembling ganglia. These lesions may be related to perimeniscal or perilabral cysts, rather than to true myxomas. Because of their juxta-articular location and ganglion-like appearance, these lesions are discussed further with synovial lesions in Chapter 9 (55).
Deep Aggressive Angiomyxoma
The term aggressive angiomyxoma was use by Steeper and Rosai in 1983 (56) to describe a distinct myxoid
neoplasm characteristically occurring in the perineal and pelvic regions of adult women (5, 56). The WHO recognized this lesion as deep aggressive angiomyxoma (6). It occurs most commonly in women in their third to sixth decade of life (peak incidence in the fourth decade), with a female-to-male ratio of more than 6:1 (5, 6, 7, 57, 58, 59). Common locations include the pelvicoperineal and retroperitoneal regions (5, 6, 7, 59). In men, the tumor involves analogous sites including the scrotum and inguinal area (60, 61). Aggressive angiomyxoma may be asymptomatic or cause pain and dyspareunia. Lesions may be mistaken for a Bartholin cyst because of their location. Tumors have been reported during pregnancy, and tumor growth during pregnancy and their prevalence in women’s reproductive years suggests that these lesions are hormone-dependent (62, 63).
neoplasm characteristically occurring in the perineal and pelvic regions of adult women (5, 56). The WHO recognized this lesion as deep aggressive angiomyxoma (6). It occurs most commonly in women in their third to sixth decade of life (peak incidence in the fourth decade), with a female-to-male ratio of more than 6:1 (5, 6, 7, 57, 58, 59). Common locations include the pelvicoperineal and retroperitoneal regions (5, 6, 7, 59). In men, the tumor involves analogous sites including the scrotum and inguinal area (60, 61). Aggressive angiomyxoma may be asymptomatic or cause pain and dyspareunia. Lesions may be mistaken for a Bartholin cyst because of their location. Tumors have been reported during pregnancy, and tumor growth during pregnancy and their prevalence in women’s reproductive years suggests that these lesions are hormone-dependent (62, 63).
KEY CONCEPTS
Aggressive angiomyxoma is a distinct myxoid neoplasm characteristically occurring in the perineal and pelvic regions of adult women.
It is rare in men, involving analogous sites including the scrotum and inguinal area.
Simple excision is usually curative without recurrence.
Radiographs are usually normal or show a nonspecific soft tissue mass with displacement of pelvic structures.
CT demonstrates a well-defined mass with attenuation lower than that of muscle.
MR will show a homogeneous or mildly heterogeneous fluid-like mass.
A characteristic swirled or layered appearance is described in greater than 80% of cases on CT and MR imaging.
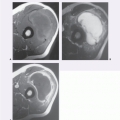
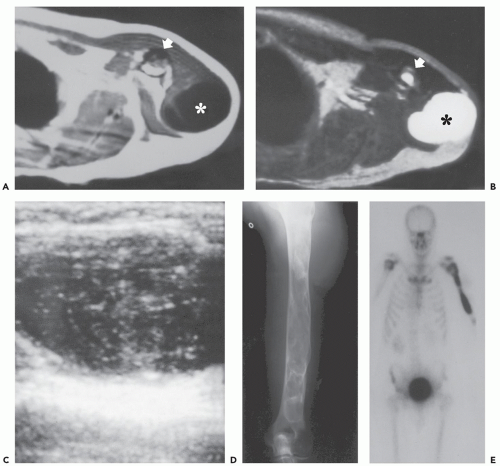 FIGURE 11.8 Myxoma and fibrous dysplasia: Mazabraud syndrome in a 55-year-old man presenting with a soft tissue mass in the shoulder. Axial T1-weighted (TR/TE; 600/17) (A) and T2-weighted (TR/TE; 2,500/90) (B) MR images of the shoulder show a well-defined, homogeneous, intramuscular mass (asterisk). The signal intensity suggests that it is cyst-like. Note the abnormal signal intensity in the marrow of the humerus (arrows). C: Ultrasound of the soft tissue mass shows the lesion not to be a simple cyst, with the appearance of a complex mass. D: Radiograph of the humerus shows changes compatible with fibrous dysplasia. E: Technetium-99m bone scan confirms that the fibrous dysplasia is monostotic and there is no radionuclide uptake in the myxoma. |
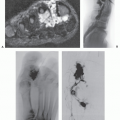
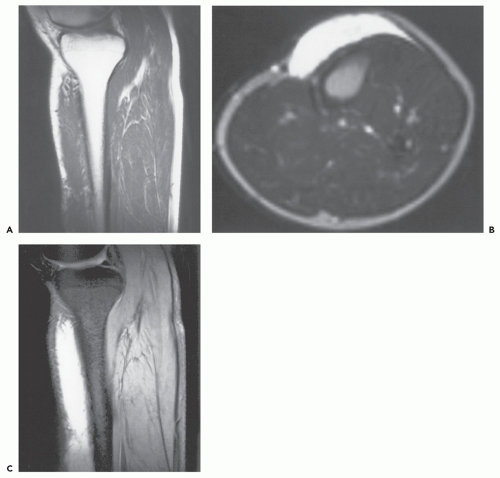 FIGURE 11.9 Subcutaneous myxoma: MR imaging features in the lower leg of a 32-year-old man. Sagittal T1-weighted (TR/TE; 600/20) (A) and axial T2-weighted (TR/TE; 2,500/90) (B) MR images show a well-defined, subcutaneous mass anterior to the tibia. The large size of the lesion precludes clearly identifying the origin in the subcutaneous tissue or aponeurosis. C: Corresponding sagittal gradient-echo image shows markedly increased signal intensity, similar to that seen on the T2-weighted MR image. (Case courtesy of Robert G. Dussault, MD, and Phoebe A. Kaplan, MD.) |
At gross pathologic evaluation, deep aggressive angiomyxomas are often quite large (10 to 20 cm in size), lobulated, partially circumscribed, gelatinous masses (5, 6, 7, 59). Microscopically, deep aggressive angiomyxomas are low to moderate in cellularity and composed of small, stellate, spindled cells (64). Scattered, dilated vessels of variable size (from capillaries to large vessels) are usually seen in the lesion (65). The vessels are often surrounded by smooth muscle cells. Mitoses are infrequent. Cytogenetic aberrations are seen with rearrangement of 12q14-15 in various translocations (5, 6, 7, 59, 66, 67).
Although wide excision is the treatment of choice for deep angiomyxoma, the large size, infiltration, and location of lesions often precludes complete removal (65, 68, 69). Thus, the local recurrence rate is generally high, ranging from 30% to 100% (5, 6, 7, 59, 65); however, more recent studies have demonstrated much lower rates of local recurrence following aggressive surgical excision (70, 71). Local recurrence is often adequately treated by reexcision alone, without the need of adjunct therapy (65). Deep aggressive angiomyxoma is generally considered to have no significant metastatic potential and documented metastases are exceedingly rare (72, 73).
Radiographs are usually normal or show a nonspecific soft tissue mass with displacement of pelvic structures. Angiography typically reveals a hypervascular mass, whereas sonography demonstrates a heterogeneous, hypoechoic, soft tissue mass (74, 75, 76, 77, 78). Internal echogenic septa, cystic areas, and a hyperechoic rim are also described (Fig. 11.10).
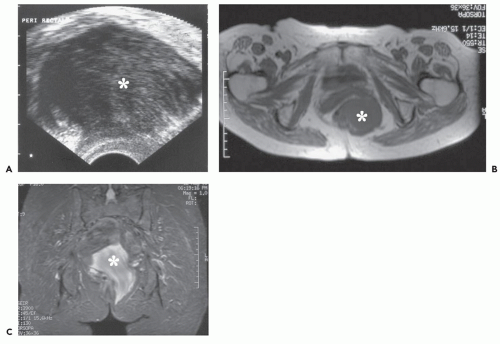 FIGURE 11.10 Deep aggressive angiomyxoma: Ultrasound and MR imaging features in a 77-year-old woman with a pelvic mass. A: Endorectal sonogram demonstrates a hypoechoic, heterogeneous, perirectal soft tissue mass (asterisk). Axial T1-weighted (TR/TE; 550/14) (B) and coronal STIR (TR/TE/TI; 3,900/45/130) (C) MR images show the perivaginal soft tissue mass (asterisk) with low signal intensity on T1-weighting and high signal on T2-weighting. There is heterogeneity on the long TR image and a swirled/layered appearance. |
CT shows a well-defined mass with attenuation lower than that of muscle (Fig. 11.11) (60, 74, 76, 77, 78, 79). On T1-weighted MR images, deep aggressive angiomyxoma is usually low to intermediate in signal intensity (60, 76, 78, 79, 80). On T2-weighted MR images, these lesions reveal intermediate-to-high signal intensity, likely because of the high water content seen histologically (Figs. 11.10 and 11.11) (76, 79, 80). Deep aggressive angiomyxoma is described as having a characteristic swirled or layered appearance on CT and MR imaging (Figs. 11.10 and 11.11). This appearance (83% of cases by CT or MR imaging) is seen on T2-weighted MR imaging and post-contrast CT or MR images. In the study by Outwater et al. (79), the strands of tissue creating this swirled architecture were often best seen on contrastenhanced CT or MR imaging, and were mildly lower signal intensity on long TR images. Cross-sectional imaging, particularly CT or MR imaging, reveals the effect of these often large masses on surrounding pelvic structures and whether or not the tumor traverses the pelvic diaphragm; anatomic features that are essential in planning surgical resection.
Myoepithelioma/Myoepithelial Carcinoma/Mixed Tumor
Myothelioma and mixed tumor of soft tissue are currently grouped together by the WHO because of their morphological and immunophenotypical similarity (81, 82, 83, 84). Myoepithelioma is composed exclusively (or predominantly) of myoepithelial cells while mixed tumors show ductal differentiation (84). About 10% of myoepitheliomas will contain a ductal component and
are then termed mixed tumor (84). Mixed tumor of soft tissue is analogous to the chondroid syringoma described in dermatopathology (Chapter 12). Lesions with large epithelioid cells showing cytoplasmic vacuolation are termed parachordoma, now considered a morphologic variant of mixed tumor/myoepithelioma (81, 84), with the designation of parachordoma used to emphasize its morphological similarities to axial chordoma (85). Malignant myoepithelial neoplasms are designated myoepithelial carcinoma (84).
are then termed mixed tumor (84). Mixed tumor of soft tissue is analogous to the chondroid syringoma described in dermatopathology (Chapter 12). Lesions with large epithelioid cells showing cytoplasmic vacuolation are termed parachordoma, now considered a morphologic variant of mixed tumor/myoepithelioma (81, 84), with the designation of parachordoma used to emphasize its morphological similarities to axial chordoma (85). Malignant myoepithelial neoplasms are designated myoepithelial carcinoma (84).
KEY CONCEPTS
Myothelioma and mixed tumor of soft tissue and are grouped together by the WHO because of their morphological and immunophenotypical similarity.
About 10% of myoepitheliomas will contain a ductal component and are then termed mixed tumor.
Mixed tumor of soft tissue is analogous to the chondroid syringoma described in dermatopathology.
The term parachordoma, is a morphologic variant of mixed tumor/myoepithelioma (81, 84), with morphological similarities to axial chordoma.
Adults are typically affected in the fourth to sixth decade of life, with an average age of 35 years.
Lesions are most frequently in the subcutaneous or deep subfascial soft tissues of the extremities; the upper extremity is more commonly affected than the lower extremity.
Imaging of these lesions is only rarely reported, showing nonspecific features on CT and MR imaging.
Cystic, necrotic, or hemorrhagic areas may be apparent.
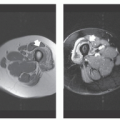
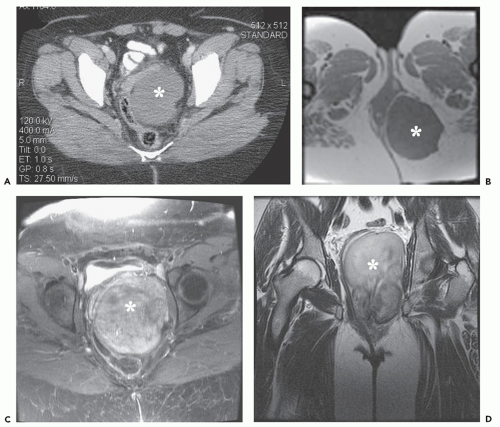 FIGURE 11.11 Deep aggressive angiomyxoma: CT and MR imaging features in a 64-year-old woman with a pelvic mass. A: CT shows a well-defined, low attenuation, soft tissue mass (asterisk) displacing the rectosigmoid portion of the colon. Multiple MR images including axial T1-weighted (TR/TE; 500/20) (B), axial post-contrast fat-suppressed T1-weighted (TR/TE; 400/15) (C), and coronal T2-weighted (TR/TE; 4,000/60) (D) reveal the perirectal soft tissue mass (asterisk). There is low attenuation on T1-weighting, moderate diffuse enhancement and intermediate signal intensity on T2-weighting. Note swirled/layered appearance after contrast and on the long TR image. |
Adults are typically affected in the fourth to sixth decade of life, with an average of 35 years of age (6, 7, 82, 83). However, as with many neoplasms, there is a wide age range, and 20% of patients are children younger than 10 years (6, 7, 82, 83, 86). A mild male predilection is reported in some series (6, 7, 82, 83, 87). Sites of involvement are most frequently the subcutaneous or deep subfascial soft tissues of the extremities (11, 12, 74, 75). The upper extremity is more commonly affected than the lower extremity. Other involved locations include the head/neck, trunk, and rarely, bone (11, 12, 74, 75). Clinically, patients present with a nonspecific painless swelling or mass.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


