Summary of clinical and imaging features of arthropathies discussed in this chapter is provided in Table 10.1.
AMYLOIDOSIS
Clinical and Pathologic Features
Amyloidosis is a systematic disorder characterized by the infiltration of various organs by a homogeneous eosinophilic material consisting of protein fibers in a ground substance of mucopolysaccharides. There are three major types of systemic amyloidosis: (1) primary amyloidosis, the most common form, in which bone marrow produces too much of certain fragments of antibody protein, which builds up in the bloodstream and deposits in the body tissues; (2) familial (hereditary) amyloidosis, which is a genetic form, because of mutations in the gene TTR, inherited in autosomal dominant manner; and (3) secondary amyloidosis, which develops secondary to certain chronic conditions such as tuberculosis or rheumatoid arthritis. Amyloid arthropathy is a sign of acquired idiopathic systemic amyloidosis and is a condition that results in noninflammatory arthropathy. All forms of amyloidosis are characterized pathologically by extracellular deposition of insoluble nonbranching beta-pleated protein fibrils formed as a result of abnormal protein synthesis. Amyloid is seen within the synovium and bone marrow as extensive extracellular deposits of brightly eosinophilic/hyaline amorphous material. Histopathologic sections stained with Congo red have a characteristic applegreen birefringence when examined under polarized light (Fig. 10.1).
Clinically, amyloidosis bears a striking resemblance to rheumatoid arthritis, because the joints are stiff and painful and the arthropathy is bilateral and symmetric. There is a predilection for large joints such as the hips, knees, shoulders, and elbows. Subcutaneous nodules are noted over the extensor surfaces of the forearm and dorsum of the hand, often mimicking the rheumatoid nodules. Another characteristic feature is the massive involvement of the soft tissues, giving the patient an almost pathognomonic appearance known as “shoulder pad sign” or “football player shoulders.” Carpal tunnel syndrome is frequently an associated abnormality.
The bone abnormalities and arthropathy associated with deposition of B2-microglobulin (B2-MG) amyloid are well-recognized complications of long-term hemodialysis and chronic renal failure. Standard dialysis membranes do not filter B2-MG, a low molecular weight serum protein. It therefore accumulates in the bones, joints, and soft tissues. Clinically, characteristic pain and decreased joint mobility occur in the affected joints.
Imaging Features
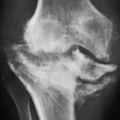
Regardless of cause, imaging studies show massive accumulation of amyloid around the joints, and there is invasion of the periarticular tissue, capsule, and joint. Also, deposits can be seen in the synovium. The articular ends of the bone can be destroyed, and both subluxations and pathologic fractures are frequently encountered. In addition, focal osteolytic lesions, particularly in the bones of the upper extremities (Fig. 10.2) and in the proximal ends of the femora, can be seen. The MRI manifestations of amyloidosis include intermediate-to-low signal intensity deposits of amyloid material in the synovium, ligaments, and tendons with or without erosive changes (Fig. 10.3). Scintigraphy using 123I-serum amyloid P component has been used to identify the systemic distribution of amyloid deposit.
Table 10.1 MISCELLANEOUS ARTHROPATHIES: CLINICAL AND IMAGING FEATURES
Amyloidosis (M > F)
Large joints (hips, knees, shoulders, elbows)
Articular and periarticular erosions, osteoporosis (periarticular), joint subluxations, pathologic fractures
Standard views of affected joints
Radionuclide bone scan (scintigraphy)
Multicentric reticulohistiocytosis (F > M)
Hands (distal and proximal interphalangeal joints)
Feet
Soft tissue swelling, articular erosions, lack of osteoporosis
Joint effusion, osteoporosis, symmetrical and concentric joint space narrowing, articular erosions, widening of intercondylar notch, squaring of patella; very similar to changes of juvenile rheumatoid arthritis
Standard views of affected joints
Magnetic resonance imaging
Figure 10.1 ▪ Pathology of amyloidosis. A: Photomicrograph depicts amyloid deposit in the bone marrow (H&E, original magnification ×10). B: Under polarized light, the amyloid deposits are birefringent and apple-green (original magnification ×10). (From Bullough PG. Atlas of Orthopedic Pathology with Clinical and Radiologic Correlation, 2nd ed. New York, NY: Gower Medical Publishing, 1992:16.2-16.4., Figs. 8.12 and 8.13, p. 8.6.)
Figure 10.2 ▪ Amyloidosis. A: Anteroposterior radiograph of the right shoulder of an 80-year-old man demonstrates a moderate degree of juxta-articular osteoporosis, soft tissue swelling, and a large osteolytic lesion in the humeral head. The glenohumeral joint space is relatively well preserved. B: Radionuclide bone scan shows an increased uptake of technetium-labeled methylene diphosphonate (MDP) around the shoulder.
Figure 10.3 ▪ MRI of amyloidosis. A: Coronal T2-weighted fat-saturated MRI of the knee in a patient with primary amyloidosis demonstrates thickening of the popliteus tendon (arrowhead) and the proximal superficial fibers of the medial collateral ligament (arrow) because of extensive deposits of intermediate signal intensity amyloid tissue. Note also amyloid deposition in the intercondylar notch. B: Sagittal T2-weighted MRI of the knee shows the hypointense synovial deposits of amyloid tissue (arrows).
Treatment
Although there is no cure for amyloidosis, therapy is directed toward relieving the symptoms and limitation of further production of amyloid protein. Treatment includes chemotherapy agents such as melphalan or cyclophosphamide and corticosteroids such as dexamethasone. Recently, other drugs such as bortezomib, thalidomide, and lenalidomide, which are a thalidomide derivative, have been tried with some promising results. In most severe cases, autologous peripheral blood stem cell transplantation using high-dose chemotherapy and transfusion of stem cells has been advocated. Surgical treatment consists of removal of affected organs followed by organ transplantation.
MULTICENTRIC RETICULOHISTIOCYTOSIS (MRH)
Multicentric reticulohistiocytosis (MRH) is a rare systemic granulomatous disorder of unknown cause seen in adulthood and is characterized by the proliferation of the histiocytes (macrophages) in the skin, the mucosa, the subcutaneous tissue, and the synovium. It was first described in 1937 as a nondiabetic cutaneous xanthomatosis. The disorder has been also called lipoid dermatoarthritis, reticulohistiocytoma, lipid rheumatism, giant cell reticulohistiocytosis, giant cell histiocytoma, and giant cell histiocytosis. Goltz and Lymon proposed the name multicentric reticulohistiocytosis for this condition in 1954 because of the multifocal origin and systemic nature of the disease. It usually begins during the fourth decade of life, and women are more commonly affected than men, with ratio 3:1. In ˜60% to 70% of patients, polyarthralgia is the first manifestation of the disease. Approximately 25% of cases have been associated with the presence of neoplasia, including all types of cancer, solid and hematologic. MRH frequently coexists with autoimmune diseases, including Sjögren syndrome, systemic lupus erythematosus, scleroderma, and dermatomyositis. Occasionally, nonautoimmune conditions have been associated with MRH, such as diabetes mellitus, thyroid disease, colitis, and dyslipidemia.
Laboratory markers are poorly sensitive and not specific for MRH. About half of the patients show elevated erythrocyte sedimentation rate and C-reactive protein. Serum autoantibodies are only occasionally detected and generally support the coexistence of an autoimmune disorder. Histopathologic findings include histiolymphocytic infiltrate within the dermis with small, sometimes multinucleated histiocytes. These cells are not foamy, which allows distinguishing them from other forms of histiocytic disorders, such as sarcoidosis or xanthogranulomatosis. More advanced dermal lesions are characterized by the pathognomonic multinucleated giant cells with a ground-glass eosinophilic cytoplasm (megalocytes). Additional findings include a strong positivity of histiocytes for periodic acid-Schiff (PAS) and expression of acid phosphatase, nonspecific esterase, and lysozyme. Triglycerides, cholesterol, and phosphate esters appear to be present in the lesion, suggesting either that histiocytes are stimulated to produce these substances or that this is a form of lipid storage disease. Immunohistologically, MRH histiocytes express positivity for tartrate-resistant acid phosphatase (TRAP), CD68, CD45, MAC387, vimentin, lysosome, and human alveolar macrophage 56 (HAM-56); however, there is conspicuous negativity for S100 protein, CD1a, and factor XIIIa. Furthermore, the synovial fluid mononuclear cells from patients with MRH express macrophage-specific markers including CD11b, CD14, CD68, and class II HLA DR. Quantitative amounts of proinflammatory cytokines are elevated, including TNF-alpha, IL-6, IL-12, and IL-1-beta. More recent data support the hypothesis that synovial fluid macrophages may differentiate into osteoclasts following RANKL pathway activation or macrophage colony-stimulating factor (M-CSF) as suggested in other inflammatory diseases. Ultrastructurally, the histiocytes resemble type A synoviocytes. The cytoplasm contains osmiophilic membrane-bound lysosomal granules, often closely associated with a single large stellate Golgi apparatus.
Figure 10.4 ▪ Multicentric reticulohistiocytosis. Clinical photograph of the hands of the patient with MRH shows characteristic erythematous nodules on the dorsal aspect of the metacarpophalangeal and interphalangeal joints.
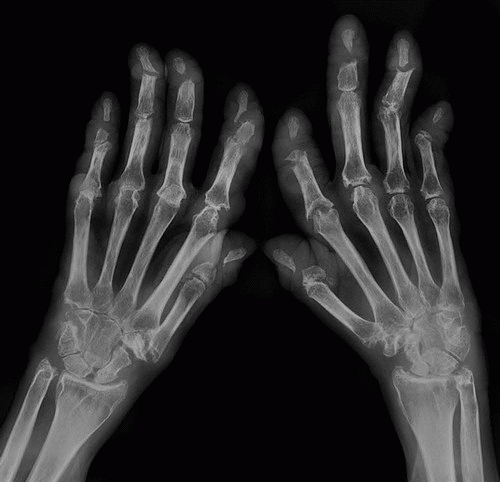
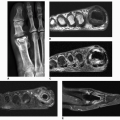
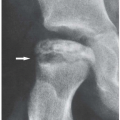
Clinical findings include characteristic cutaneous manifestations appearing as translucent brown-reddish to fleshcolored papulonodular lesions of variable sizes (1 mm to 1 cm or larger) (Fig. 10.4). The other clinical features may be very similar to those of rheumatoid arthritis, consisting of soft tissue swelling, stiffness, and tenderness of the joints. Unlike rheumatoid arthritis, however, the distal interphalangeal joints are most frequently affected. Less commonly affected are the proximal interphalangeal, metacarpophalangeal, shoulder, and elbow joints. Occasionally, the articular lesions may be marked by severe destruction similar to arthritis mutilans of rheumatoid arthritis or psoriatic arthritis (Figs. 10.5 and 10.6). The characteristic absence of significant periarticular osteoporosis distinguishes this disorder from the inflammatory arthritides, and there is also no periosteal new bone formation, which distinguishes it from psoriatic arthritis or juvenile idiopathic arthritis. Lack of osteophytes and interphalangeal ankylosis, and the presence of soft tissue nodules and atlantoaxial abnormalities including subluxation and erosion of the odontoid process distinguish this arthropathy from erosive osteoarthritis. At times, the pattern of bone erosions with sclerotic margins and overhanging edges, and the presence of soft tissue masses, may mimic those of gout (Fig. 10.7). Unlike gout, however, there is symmetrical distribution of the lesions in the hands and feet and lack of calcification within soft tissue nodules.
Figure 10.5 ▪ Multicentric reticulohistiocytosis. Dorsovolar radiograph of both hands of a 57-year-old woman with long-standing polyarthralgia, soft tissue swelling, and deformities of the fingers demonstrates severe destruction of multiple carpometacarpal, metacarpophalangeal, and interphalangeal joints similar to those seen in rheumatoid or psoriatic arthritis.
Treatment consists of systemic steroids, cytotoxic drugs such as cyclophosphamide, chlorambucil, methotrexate, leflunomide, and infliximab. Bisphosphonates such as alendronate and zoledronate have been reported to improve skin lesions and arthritis.
Figure 10.6 ▪ Multicentric reticulohistiocytosis. Dorsovolar radiograph of both hands of a 63-year-old man shows arthritis mutilans affecting mainly distal interphalangeal joints.
Figure 10.7 ▪ Multicentric reticulohistiocytosis. A: A 46-year-old woman presented with distal interphalangeal joints pain and soft tissue swelling. Note sharply marginated erosions at the distal interphalangeal joints (arrows) resembling gout. B: Radiograph of the fingers of the right hand of the 65-year-old woman shows small erosions at the distal interphalangeal joints of the index and middle fingers (arrowheads) associated with soft tissue masses (arrows) resembling gouty tophi.
SARCOIDOSIS
Sarcoidosis is a systemic inflammatory disorder predominantly affecting young adults, characterized by the presence of noncaseating granulomas in affected organs. The disease has a worldwide distribution, with the greatest incidence in Sweden. Although the etiology still remains unsettled, the evidence that sarcoidosis most commonly involves the lungs, eyes, and skin, focused the search for environmental causes such as exposures to airborne antigens. In fact, some of the earliest studies have reported association of sarcoidosis with exposures irritants found in rural settings, such as emissions from wood-burning stoves and tree pollen. More recently, association of sarcoidosis with exposure to inorganic particles, insecticides, and moldy environments has been suggested. Currently, the investigators found compelling evidence to support the hypothesis implying certain environmental foreign nonparticulates as a plausible cause of this condition in individuals with a genetically based immune dysregulational predisposition. Some researchers suggested that a predisposition to acute sarcoid arthritis is carried by the HLA DQ2-DR3 haplotype that appears to be transmitted as a dominant genetic trait. Many genetic associations have been linked sarcoidosis with genes within the major histocompatibility complex (MHC) locus. Most recent studies suggested the novel gene BTNL2 (butyrophilin-like) has been associated with sarcoidosis of Caucasian patients. The active granulomatous inflammation is associated with a dominant expression of T-helper (Th) 1 cytokines (interferon gamma [IFN-gamma]), interleukin-12 and IL-18, and tumor necrosis factor (TNF).
Only gold members can continue reading. Log In or Register to continue