Neuroradiology: Brain
Timothy J. Amrhein
Usman Manzoor
Zoran Rumboldt
The authors and editors acknowledge the contribution of the Chapter 6A author from the third edition: Christopher K. Moses, MD.
Case 6.1
HISTORY: A 3-year-old boy with seizures
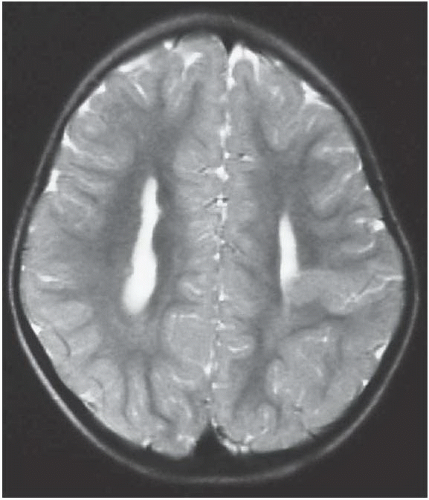 FIGURE 6.1.1 |
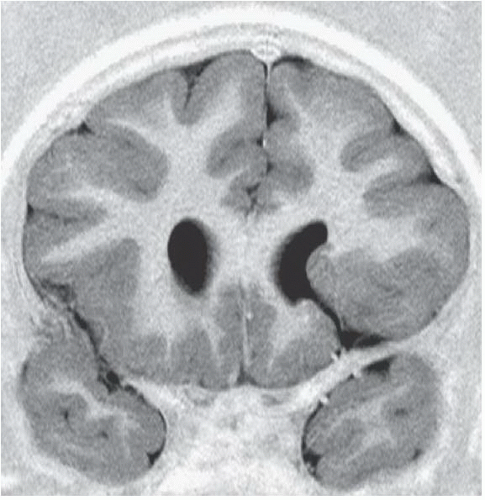 FIGURE 6.1.2 |
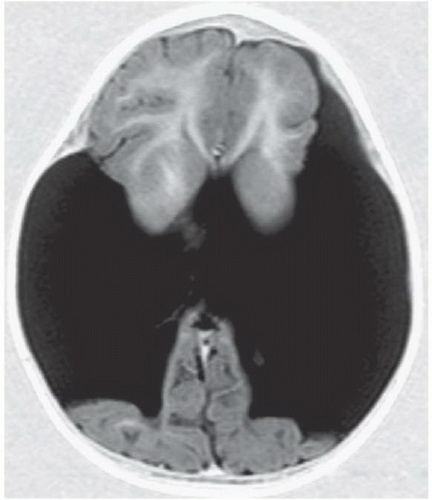 FIGURE 6.1.3 |
FINDINGS: Axial T2-weighted (Fig. 6.1.1) MR image demonstrates gray matter extending from the surface of the brain to the ependymal lining of the left lateral ventricle. Coronal T1-weighted inversion recovery image from a different patient (Fig. 6.1.2) demonstrates a gray matter-lined cleft extending from the brain surface to the left lateral ventricle. Axial T1 inversion recovery MR image in another patient (Fig. 6.1.3) demonstrates wide bilateral cerebrospinal fluid (CSF) clefts indicative of open-lip schizencephaly.
DIAGNOSIS: Schizencephaly (closed-lip type)
DISCUSSION: Schizencephaly may be close-lipped or open-lipped, unilateral or bilateral, and typically manifests with seizures and mental retardation. It is a disorder of neuronal migration caused by second-trimester insult or genetics resulting in a full-thickness communication between the brain surface (pia) and ventricular system ependyma. The cleft is lined by abnormal gray matter, which may act as a seizure focus. If the abnormal gray matter is closely apposed, the disorder is close-lipped; if unopposed, it is open-lipped.
The diagnosis is obvious in open-lipped schizencephaly—a wide CSF communication exists between the lateral ventricle and the subarachnoid space. Close-lipped schizencephaly is more subtle, and the key imaging feature is ectopic gray matter extending from the ventricular wall to the brain surface and outlining a thin, sometimes imperceptible cleft. Other clues to the diagnosis include gyri radiating into the cleft, associated subependymal heterotopias, and a dimple in the wall of the lateral ventricle. Multiplanar imaging is required, because isolated imaging in just one plane may miss the defect. Associated anomalies include absence of the septum pellucidum (90%) and septo-optic dysplasia (1,2).
Aunt Minnie’s Pearls
Close-lipped schizencephaly can be subtle—the key diagnostic feature is ectopic gray matter extending from the lateral ventricle to the brain surface outlining a thin cleft.
Multiplanar imaging is required in any child presenting with seizures to avoid the pitfall of missed schizencephaly.
Case 6.2
HISTORY: A 15-month-old girl with seizures
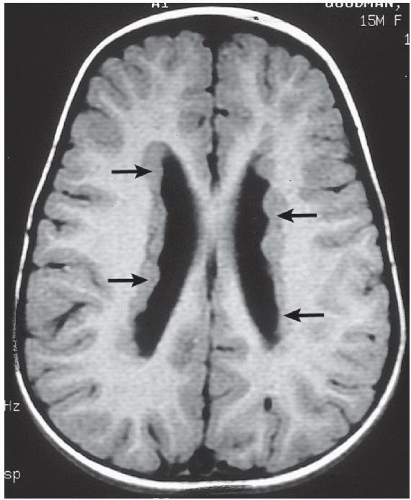 FIGURE 6.2.1 |
 FIGURE 6.2.2 |
 FIGURE 6.2.3 |
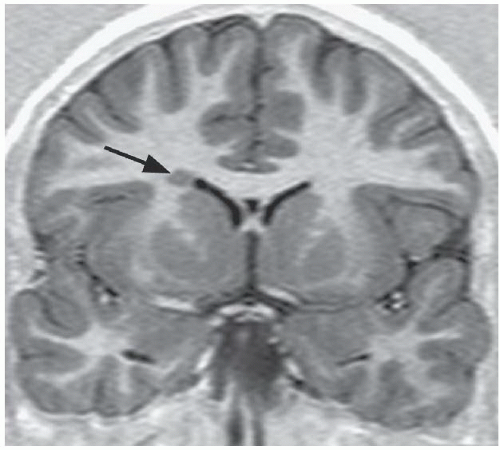 FIGURE 6.2.4 |
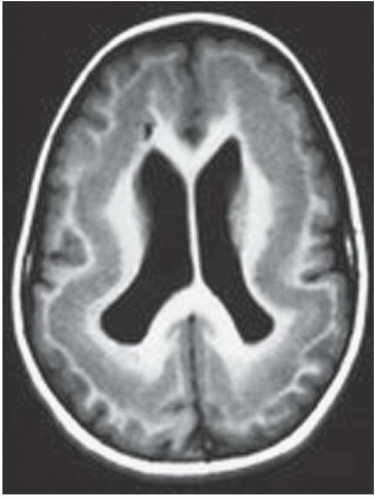 FIGURE 6.2.5 |
FINDINGS: Axial T1-weighted (Fig. 6.2.1) and T2-weighted (Fig. 6.2.2) MR images show diffuse subependymal nodules (arrows) lining both lateral ventricles. The nodules maintain isointensity with cortical gray matter on both pulse sequences. Coronal T1-weighted inversion recovery MR image in a different patient (Fig. 6.2.3) demonstrates large areas of gray-matter signal adjacent to the bilateral lateral ventricles (white arrows), consistent with multiple foci of gray-matter heterotopia. A coronal T1-weighted inversion recovery MR image from a third patient (Fig. 6.2.4) demonstrates a more subtle small focus of gray-matter heterotopia adjacent to the right lateral ventricle (arrow). Axial T1 inversion recovery MR image in a different patient (Fig. 6.2.5) demonstrates a band of tissue with signal characteristics of gray matter within the white matter between the cortex and the lateral ventricles, compatible with band heterotopia.
DIAGNOSIS: Gray-matter heterotopia
DISCUSSION: Gray-matter heterotopia is a relatively common congenital abnormality that results from an in utero arrest of neuronal migration. The abnormally located gray matter is usually found in the subependymal region of the lateral ventricles, around the trigones in particular. Occasionally, the heterotopias are more peripherally located near the cerebral cortex. Patients come to clinical attention because of seizures and developmental delay.
Heterotopias may be classified into three groups: subependymal heterotopia, focal subcortical heterotopia, and diffuse (band) heterotopia. The imaging hallmark is the isointensity with cortical gray matter on all imaging sequences and the lack of contrast enhancement. These characteristics allow the radiologist to distinguish this abnormality from the subependymal nodules that occur in tuberous sclerosis (1,3,4).
Aunt Minnie’s Pearl
To diagnose gray-matter heterotopia, the abnormalities must follow the signal intensity of gray matter on all pulse sequences and show no post-contrast enhancement.
Case 6.3
HISTORY: Fetal MR performed for prenatal ultrasound abnormality
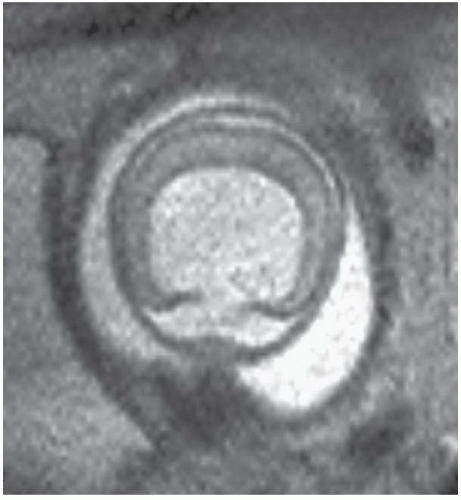 FIGURE 6.3.1 |
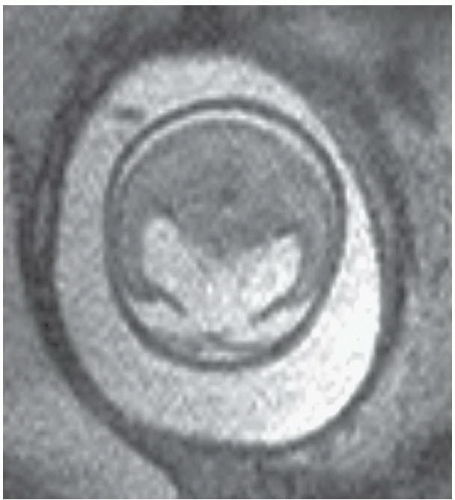 FIGURE 6.3.2 |
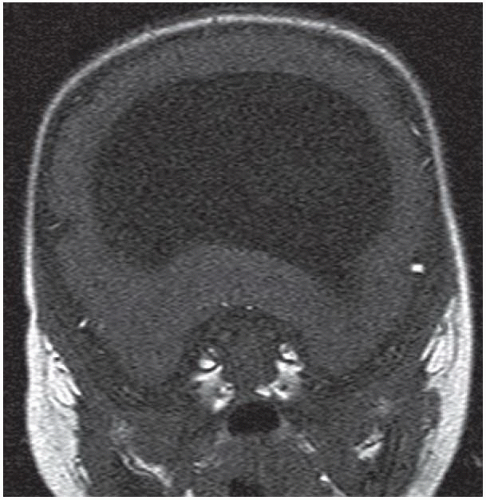 FIGURE 6.3.3 |
 FIGURE 6.3.4 |
FINDINGS: Axial single-shot T2-weighted fetal MR images (Figs. 6.3.1 and 6.3.2) demonstrate a large monoventricle and fused cerebral hemispheres. Similar findings are identified on a coronal T1-weighted MR image in a neonate (Fig. 6.3.3). Axial T2-weighted MR image in a different patient (Fig. 6.3.4) demonstrates fused thalami and basal frontal lobes but separate occipital horns of the ventricular system and more posterior portions of cerebral hemispheres indicating lobar holoprosencephaly.
DIAGNOSIS: Alobar holoprosencephaly
DISCUSSION: Holoprosencephaly refers to a continuum of congenital malformations occurring from failure of induction of the prosencephalon and absent or incomplete cleavage of the brain into distinct cerebral hemispheres. Three subtypes have been proposed according to the severity of cerebral and facial anomalies. These include alobar, semilobar, and lobar holoprosencephaly. A rare middle interhemispheric variant has also been described, wherein the cerebral hemispheres fail to divide in the posterior frontal and parietal regions. In the most severe subtype, alobar holoprosencephaly, there is complete or near complete lack of hemispheric cleavage. A primitive midline monoventricle, fused thalami, absence of the falx cerebri, and interhemispheric fissure are the typical imaging features. Midline facial anomalies ranging from single maxillary central incisor to hypotelorism to cyclopia are usually present. A multitude of extracranial and chromosomal abnormalities may occur in association with alobar holoprosencephaly, frequently resulting in a stillborn infant or death in the neonatal period (1,5,6).
Aunt Minnie’s Pearl
Midline monoventricle with fused frontal lobes and thalami = alobar holoprosencephaly.
Case 6.4
HISTORY: Fetal MR performed for prenatal ultrasound abnormality
 FIGURE 6.4.1 |
 FIGURE 6.4.2 |
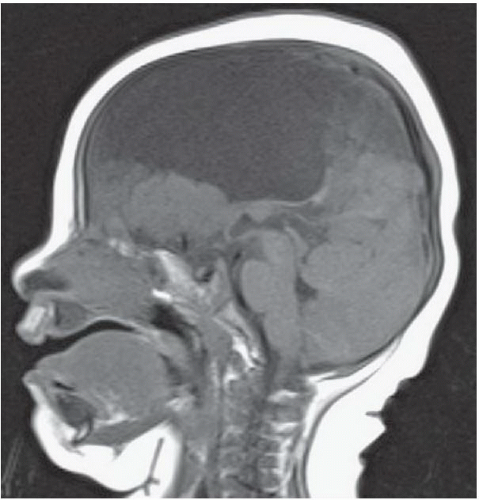 FIGURE 6.4.3 |
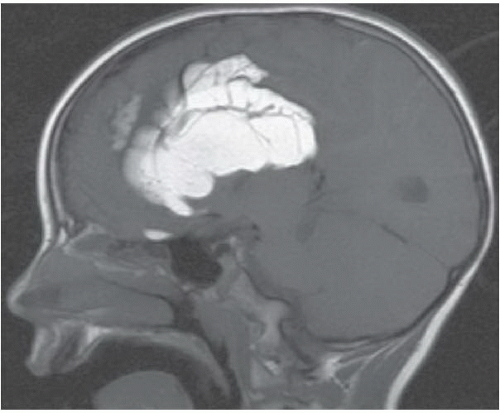 FIGURE 6.4.4 |
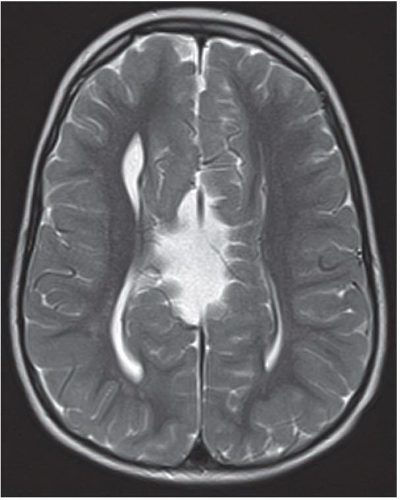 FIGURE 6.4.5 |
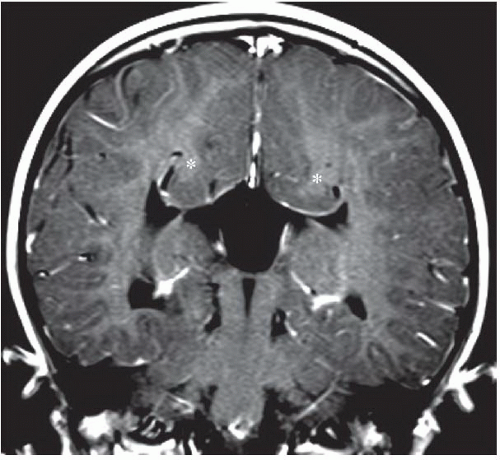 FIGURE 6.4.6 |
FINDINGS: Axial and coronal T2-weighted fetal MR image (Figs. 6.4.1 and 6.4.2) demonstrate absence of the corpus callosum and an associated interhemispheric cyst. There is also a focus of gray-matter heterotopia (arrow). Sagittal T1-weighted MR image in a different patient (Fig. 6.4.3) demonstrates a large interhemispheric cyst and absence of the corpus callosum. A sagittal T1-weighted image in yet another patient (Fig. 6.4.4) demonstrates a large high-signal-intensity mass representing a lipoma and absence of the corpus callosum. Finally, an axial T2-weighted image (Fig. 6.4.5) in a fourth patient demonstrates characteristic parallel configuration of the bilateral lateral ventricles. Coronal post-contrast T1-weighted image (Fig. 6.4.6) in the same patient reveals impression on the medial aspects of the bilateral lateral ventricles by abnormally configured white-matter tracts called Probst bundles (asterisks).
DIAGNOSIS: Dysgenesis of the corpus callosum with associated interhemispheric cyst
DISCUSSION: This case demonstrates dysgenesis of the corpus callosum with an associated interhemispheric cyst. The corpus callosum develops in an anterior-to-posterior fashion, with the posterior genu and anterior body forming first, followed by the posterior body, splenium, and rostrum. Abnormalities of the corpus callosum are often associated with other midline abnormalities such as cysts, lipomas, or abnormal anterior or hippocampal commissures. In approximately 75% of cases, callosal abnormalities are also associated with other malformations of cortical development such as heterotopia or abnormalities of sulcation; therefore it is important to carefully evaluate the brain when an abnormality of the corpus callosum is detected. The majority of patients with corpus callosum dysgenesis have decreased volume of white matter (7,8). Complete agenesis of the corpus callosum results in the development of abnormal parallel white-matter tracts called Probst bundles that run anteroposteriorly along the medial aspect of the bilateral lateral ventricles. Further, the lateral ventricles are often widely spaced and parallel in configuration.
Aunt Minnie’s Pearl
Dysgenesis of the corpus callosum is often associated with other midline abnormalities, such as cysts and lipomas, as well as other cortical abnormalities.
Case 6.5
HISTORY: A 17-year-old female with headache and vomiting
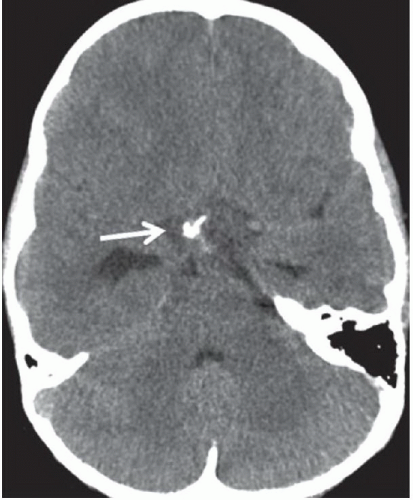 FIGURE 6.5.1 |
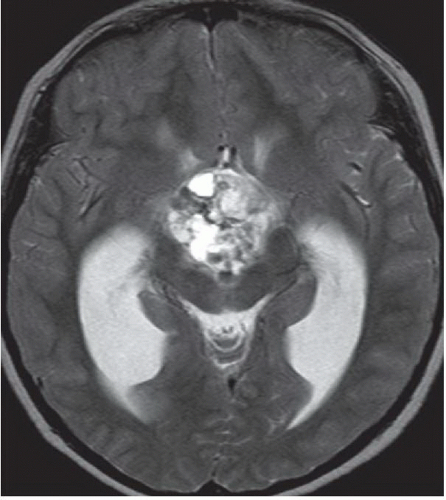 FIGURE 6.5.2 |
 FIGURE 6.5.3 |
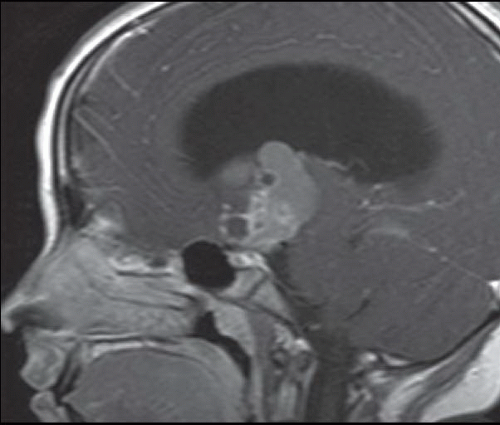 FIGURE 6.5.4 |
FINDINGS: Non-contrast axial CT image reveals a heterogeneous calcified suprasellar mass (white arrow) with solid and cystic components as well as associated hydrocephalus (Fig. 6.5.1). Axial T2-weighted image demonstrates the multilobulated nature of this mass, which contains areas of T2 hypointensity that correspond to regions of calcification identified on CT (Fig. 6.5.2). On a non-contrast sagittal T1-weighted image, the mass contains varying signal intensities ranging from isointense to adjacent gray matter to hyperintense (Fig. 6.5.3). Post-contrast T1-weighted sagittal image demonstrates a predominantly peripheral enhancement (Fig. 6.5.4).
DIAGNOSIS: Craniopharyngioma
DISCUSSION: Craniopharyngiomas are benign tumors that arise from epithelial remnants and ectopic embryonic cell rests of the Rathke’s pouch. They are the most common type of non-glial-based intracranial neoplasm in the pediatric population and exhibit a bimodal age distribution with most cases occurring either between 5 and 15 years or after 50 years of age. Clinical symptoms are usually insidious owing to the relatively slow rate of tumor growth. The most common presenting symptoms are early morning headache, vomiting, visual field defects, and endocrine abnormalities.
Craniopharyngiomas are most commonly located within the suprasellar cistern with inferior extension into the sella. However, they can be located within the suprasellar space alone, the sella in isolation, or even infrasellar.
There are two distinct histopathologic subtypes of craniopharyngiomas: adamantinomatous and squamous-papillary. Adamantinomatous craniopharyngiomas occur in children (and occasionally adults) and demonstrate the following characteristic imaging features: cyst formation, calcification, and enhancement. On MRI, cystic components are usually multiple, clustered, and exhibit varied signal characteristics dependent on their relative protein content—with greater concentrations of protein resulting in increased T1 signal and concomitant T2 hypointensity. Typically, the cysts are hyperintense to CSF on fluid-attenuated inversion recovery (FLAIR) sequences. Solid components of the mass are usually isointense to brain parenchyma on T1-weighted images, iso- to hyperintense on T2-weighted images, and demonstrate heterogeneous enhancement. One may identify peripheral enhancement of the cystic components as well. On CT, the cystic components exhibit hypoattenuation. CT is very sensitive for the detection of areas of calcification (present in about 80% of cases).
The squamous-papillary subtype occurs in adults and is primarily solid with occasional mixed solid and cystic components. The cysts in this subtype are predominantly T1 hypointense and calcifications are much less common (9,10).
Aunt Minnie’s Pearls
Suprasellar cystic and solid mass with calcifications on CT.
Characteristic variations in signal intensity of multiple cysts.
Case 6.6
HISTORY: A 5-year-old girl with short stature, delayed dentition, delayed skeletal maturation, and endocrine abnormalities
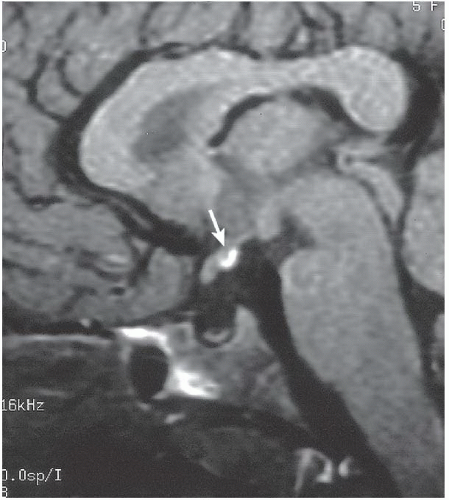 FIGURE 6.6.1 |
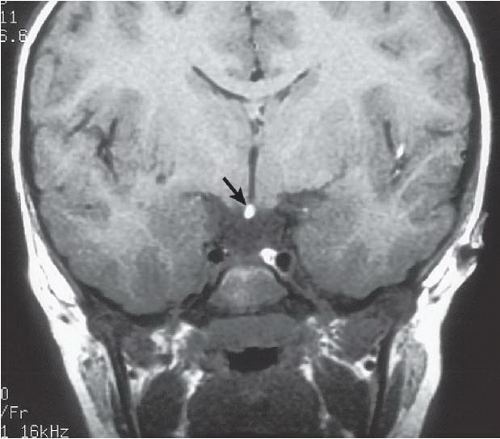 FIGURE 6.6.2 |
 FIGURE 6.6.3 |
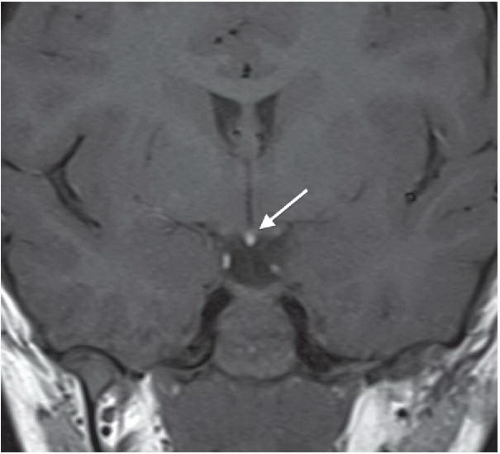 FIGURE 6.6.4 |
FINDINGS: Midline bright signal (arrow) is present in the region of the tuber cinereum on midline sagittal (Fig. 6.6.1) and coronal (Fig. 6.6.2) T1-weighted MR images. A small sella turcica, diminutive pituitary gland, and atretic pituitary stalk also are demonstrated. A second case from a different patient reinforces the same findings (Figs 6.6.3 and 6.6.4).
DIAGNOSIS: Primary panhypopituitarism with translocation of the pituitary bright spot (i.e., ectopic posterior pituitary lobe)
DISCUSSION: The posterior pituitary bright spot is thought to result from the inherent signal intensity of neurosecretory granules in the neurohypophysis; therefore, an ectopic bright spot may be found in any process that disrupts the transport of antidiuretic hormone from the hypothalamus to the posterior pituitary lobe. Primary panhypopituitarism occurs when the pituitary is congenitally hypoplastic or absent. In the clinical setting of panhypopituitarism, a high correlation exists between the hormonal disorder and ectopia of the neurohypophysis. Growth disturbances may dominate the clinical picture, as in the index case. Many patients have a history of traumatic or breech delivery, with the presumed association being perinatal rupture of the pituitary infundibulum. Characteristic MR findings of primary panhypopituitarism include a small sella turcica, diminutive pituitary gland, atretic pituitary stalk, and ectopic posterior pituitary bright spot (11,12).
Aunt Minnie’s Pearls
An ectopic posterior pituitary lobe arises from disruption of the normal transport of neurosecretory granules from the hypothalamus to the posterior pituitary.
Characteristic MR findings of primary panhypopituitarism include a small sella turcica, diminutive pituitary gland, atretic pituitary stalk, and ectopic posterior pituitary bright spot.
Case 6.7
HISTORY: A 14-month-old male with macrocephaly, developmental delay, and seizures
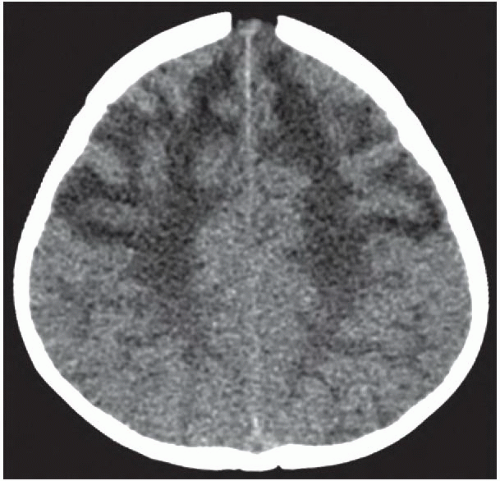 FIGURE 6.7.1 |
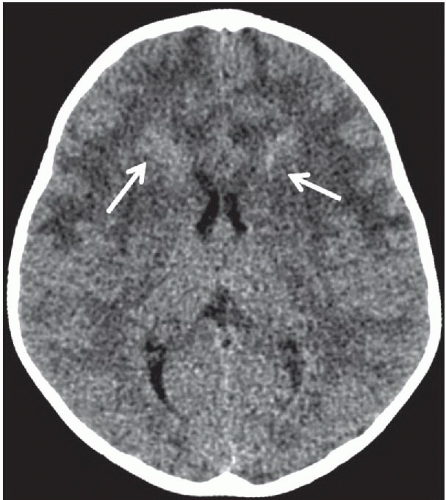 FIGURE 6.7.2 |
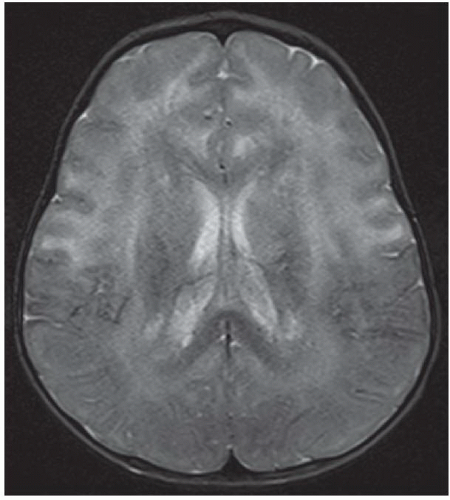 FIGURE 6.7.3 |
 FIGURE 6.7.4 |
FINDINGS: Non-contrast CT of the brain demonstrates symmetric diffuse hypoattenuation of the bilateral frontal lobe white matter and bilateral caudate heads (Fig. 6.7.1) along with hyperdense “caps” along the bilateral frontal horns (Fig. 6.7.2, white arrows). MRI reveals symmetric T1 hypointensity and T2 hyperintensity within the bilateral frontal lobe white matter (in the regions of hypoattenuation on CT) with characteristic involvement of the subcortical U fibers as well as of the deep white-matter tracts (Fig. 6.7.3). There is mild T2 hypointensity and T1 hyperintensity of the periventricular white-matter rim anterior to the bilateral frontal horns. Post-contrast images demonstrate intense enhancement along the tips of bilateral frontal horns (Fig. 6.7.4, white arrows).
DIAGNOSIS: Alexander’s disease (infantile form)
DISCUSSION: Alexander’s disease (or fibrinoid leukodystrophy) is a rare disorder characterized by a mutation to the gene for glial fibrillary acidic protein (GFAP) that leads to massive deposition of Rosenthal fibers within astrocytes resulting in demyelination and rarefaction of the subependymal, subpial, and perivascular white matter with a frontal predominance. There are three distinct clinical subgroups: infantile, juvenile, and adult. The infantile form is most common and is characterized by early onset of macrocephaly, developmental delay, and seizures. Rapid progression of the disease typically leads to death within the first 2 to 3 years of life. Macrocephaly and early onset of the clinical prodrome in combination with specific imaging findings can be diagnostic in most cases. However, establishing a definite diagnosis typically requires brain biopsy or autopsy.
Characteristic imaging findings in the infantile form of Alexander disease include a predilection for the bilateral frontal lobe white matter, which manifests as hypoattenuation on CT and corresponding T2 hyperintensity on MRI. Involvement progresses posteriorly to involve the parietal white matter as well as the internal and external capsules. Not uncommonly, similar signal abnormalities can be identified within the bilateral caudate heads. The characteristic finding is T1 hyperintensity and T2 hypointensity of the periventricular frontal rim, which is hyperdense on CT and shows avid enhancement. Further distinguishing features include involvement of the subcortical U fibers early in the disease process. Alexander disease is one of the few metabolic disorders that exhibits abnormal enhancement. Involvement of brainstem and cervical cord is uncommon. Cysts may develop in affected regions of the brain in later stages of the disease (13,14).
Aunt Minnie’s Pearls
Frontal predominant white matter involvement without sparing of the subcortical U fibers.
Involvement of the caudate heads.
T1 hyperintensity, T2 hypointensity, CT hyperdensity, and enhancement within the periventricular white-matter rim anterior to the bilateral frontal horns.
Case 6.8
HISTORY: A 16-year-old male with long-standing history of partial complex seizures
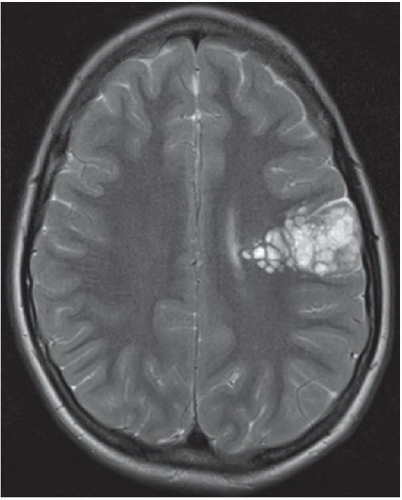 FIGURE 6.8.1 |
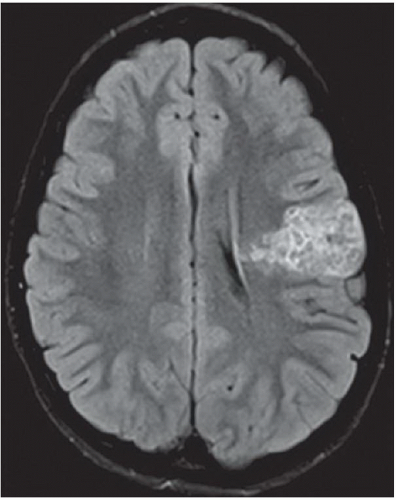 FIGURE 6.8.2 |
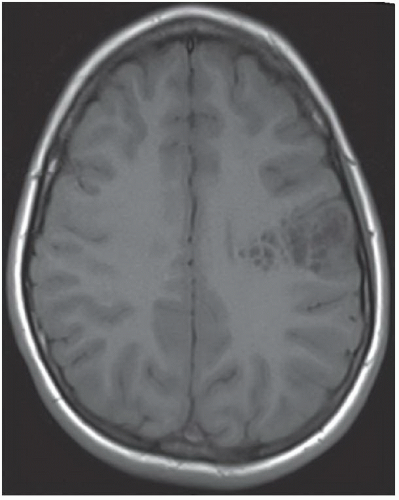 FIGURE 6.8.3 |
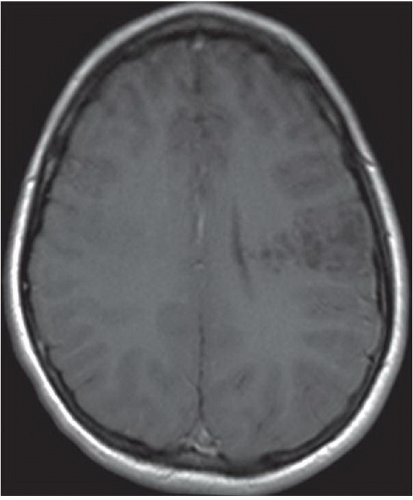 FIGURE 6.8.4 |
 FIGURE 6.8.5 |
FINDINGS: Axial T2-weighted and FLAIR images reveal a well-demarcated, multilobulated, and markedly hyperintense lesion in the posterior left frontal lobe with multiple internal cystic appearing areas, giving it a “bubbly” appearance (Figs. 6.8.1 and 6.8.2). The mass is hypointense on the T1-weighted sequence and does not enhance after the administration of contrast (Figs. 6.8.3 and 6.8.4). The mass is wedge or triangular in shape with the majority centered in the cortex and the apex extending into the adjacent subcortical white matter pointing toward the ventricle. There is no associated mass effect or surrounding vasogenic edema. An axial T2-weighted image in a second patient demonstrates a mass with similar characteristics in the right occipital lobe (Fig. 6.8.5).
DIAGNOSIS: Dysembryoplastic neuroepithelial tumor
DISCUSSION: Dysembryoplastic neuroepithelial tumor (DNET, DNT) is a benign (WHO grade I), mixed glial neuronal tumor arising from the supratentorial cortex. The most common clinical presentation is that of long-standing, drug-resistant partial complex seizures in a child or a young adult. Although DNETs can occur in any part of the supratentorial cortex, they are most commonly found in the temporal lobe. Treatment requires surgical resection, which can be curative, even if incomplete.
Imaging findings are characteristic and can be diagnostic in many cases. DNETs are classically well-demarcated, multilobulated, multicystic appearing T2 hyperintense masses that arise from the cortex. Uniquely characteristic properties of DNETs are their wedge or triangular shape with the apex pointing toward the ventricles and internal “bubbly” appearance. In general, there is minimal to no associated mass effect or surrounding vasogenic edema. DNETs typically do not enhance after contrast administration; however, in around a third of cases there may be focal punctuate or peripheral enhancement. They also show high apparent diffusion coefficient (ADC) values and low relative cerebral blood volume (rCBV) on perfusion imaging. As the tumor is slow growing, it can cause remodeling or scalloping of the inner table of the adjacent calvarium in approximately 40% to 60% cases, which is best depicted on CT. The DNETs have a strong association with focal cortical dysplasia (15,16).
Aunt Minnie’s Pearl
Cortically based, wedge shaped, multilobulated, “bubbly” lesion without mass effect in a young patient with long-standing history of seizures.
Case 6.9
HISTORY: A 14-year-old boy with seizure disorder
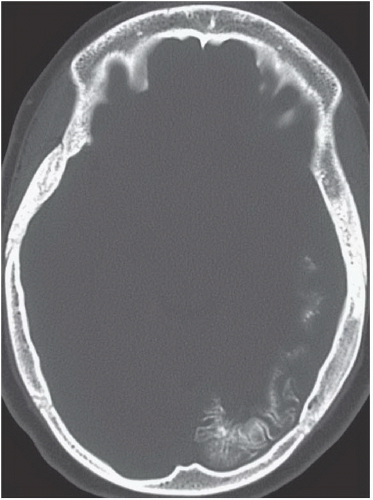 FIGURE 6.9.1 |
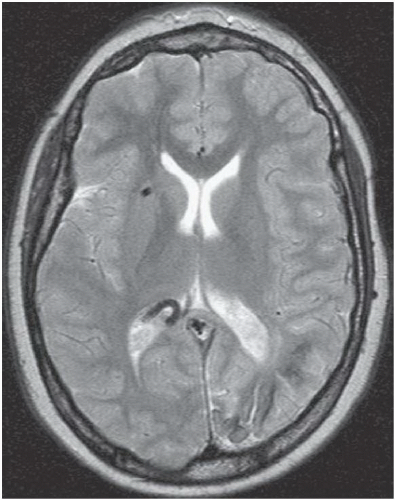 FIGURE 6.9.2 |
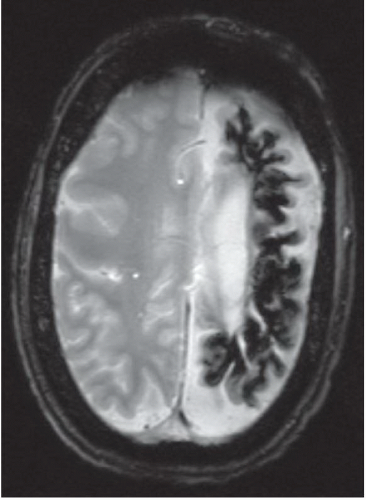 FIGURE 6.9.3 |
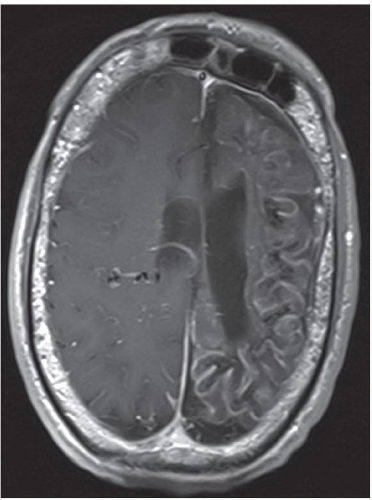 FIGURE 6.9.4 |
FINDINGS: Axial unenhanced CT scan (Fig. 6.9.1) demonstrates gyriform cortical calcifications in the temporal and occipital lobes. Axial T2-weighted MR image (Fig. 6.9.2) demonstrates gyriform low signal corresponding to the calcifications seen on the CT and volume loss of the left hemisphere, mostly within the occipital lobe. Incidentally seen are two right-sided developmental venous anomalies. Axial T2*-weighted gradient echo image in a different patient demonstrates severe atrophy of the left hemisphere with diffuse hypointensity and magnetic susceptibility artifact throughout the cortex (Fig 6.9.3). Corresponding post-contrast T1-weighted image demonstrates extensive leptomeningeal enhancement throughout the left hemisphere (Fig 6.9.4). Note the compensatory enlargement of the left frontal sinuses.
DIAGNOSIS: Sturge-Weber syndrome (i.e., encephalotrigeminal angiomatosis)
DISCUSSION: Sturge-Weber syndrome is a sporadically occurring, neurocutaneous syndrome.
The hallmark of this disease is the vascular angiomatous lesion (i.e., port-wine stain or nevus flammeus) involving the face in the distribution of the trigeminal nerve and ipsilateral brain and meninges. Imaging findings include cortical calcifications, lobar atrophy with secondary calvarial changes (i.e., Dyke-Davidoff-Masson syndrome), leptomeningeal enhancement, choroid plexus angiomas, and venous abnormalities. Evaluation of patients with suspected Sturge-Weber syndrome should include contrast-enhanced MRI because the full extent of the cortical vascular lesions may not be apparent on unenhanced imaging (1,17,18).
Aunt Minnie’s Pearls
Unilateral cortical atrophy and calcifications associated with enhancing leptomeningeal and choroid plexus angiomas = Sturge-Weber syndrome.
MRI with contrast is necessary to evaluate the full extent of the disease.
Case 6.10
HISTORY: A 21-year-old man with deafness and a recent history of double vision
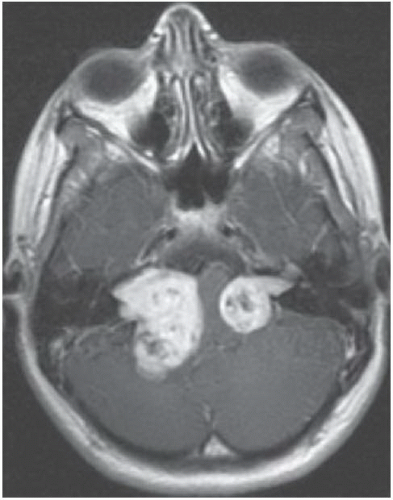 FIGURE 6.10.1 |
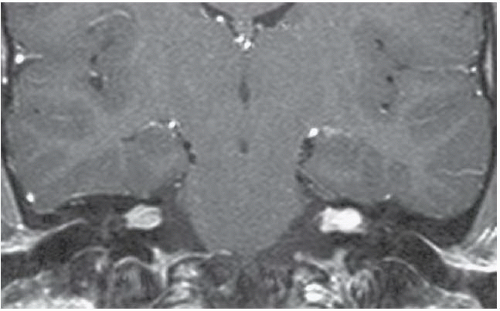 FIGURE 6.10.2 |
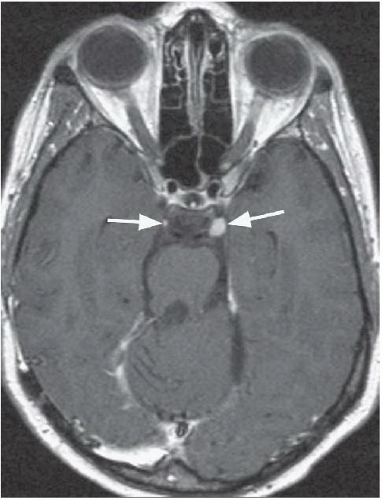 FIGURE 6.10.3 |
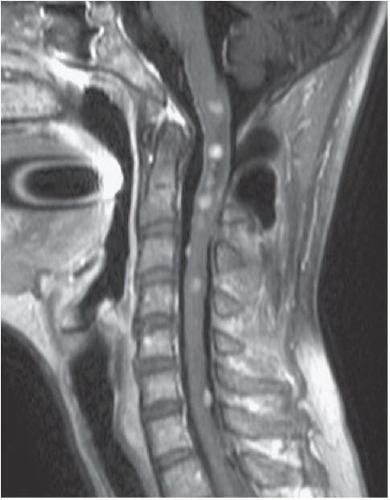 FIGURE 6.10.4 |
FINDINGS: Axial, T1-weighted post-contrast MR image (Fig. 6.10.1) demonstrates large bilateral avidly enhancing cerebellopontine angle masses with extension into the internal auditory canals. Coronal, T1-weighted post-contrast image in a different patient (Fig. 6.10.2) demonstrates bilateral enhancing internal auditory canal masses, compatible with vestibular schwannomas. Axial, T1-weighted post-contrast MR image in the same patient (Fig. 6.10.3) demonstrates bilateral enhancing masses (arrows) of the oculomotor nerves, left greater than right, compatible with schwannomas. Sagittal post-contrast T1-weighted image in the same patient (Fig. 6.10.4) demonstrates multiple central intramedullary nodules with avid enhancement, compatible with ependymomas. Additional enhancing extramedullary nodules are also seen representing meningiomas and schwannomas.
DIAGNOSIS: Neurofibromatosis type 2
DISCUSSION: Neurofibromatosis type 2 (NF 2) is an autosomal dominant disorder that is recognized as a distinct form of disease separated genetically, clinically, and radiographically from neurofibromatosis type 1 (NF 1). The incidence of NF 2 is approximately 1 case in 50,000 live births, compared with 1 case per 2,000 to 3,000 live births for NF 1. Bilateral vestibular schwannomas (also known as acoustic neuromas) are the hallmark of this disease, commonly manifesting during or soon after puberty. Schwannomas of other cranial and spinal nerves, intracranial and spinal meningiomas, and spinal ependymomas may also be found in these patients (1,19,20).
Aunt Minnie’s Pearls
Bilateral vestibular schwannomas = neurofibromatosis type 2.
NF 2 comprises multiple inherited schwannomas, meningiomas, and ependymomas (MISME).
Case 6.11
HISTORY: A 17-year-old girl with mental retardation and seizures
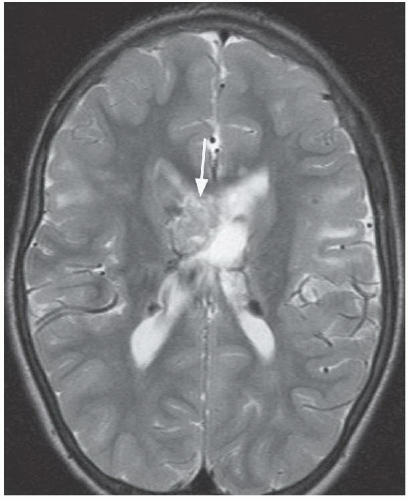 FIGURE 6.11.1 |
 FIGURE 6.11.2 |
 FIGURE 6.11.3 |
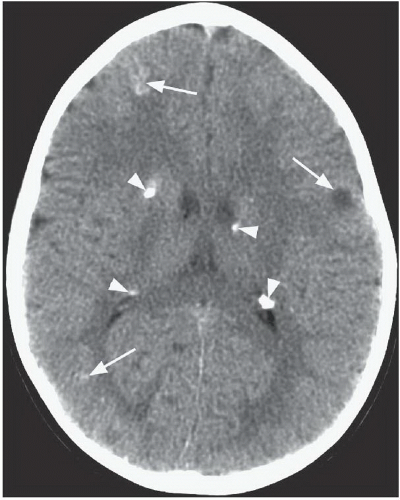 FIGURE 6.11.4 |
FINDINGS: Axial T2-weighted MR image (Fig. 6.11.1) demonstrates a large, heterogeneous mass (arrow) in the region of the foramen of Monro. Multiple low-signal-intensity foci are present along the subependymal surface of the lateral ventricles, and multiple foci of increased signal are seen in the subcortical white matter of the frontal lobes bilaterally. Axial post-contrast T1-weighted MR image (Fig. 6.11.2) demonstrates heterogeneous enhancement of the mass in the region of the foramen of Monro. Axial FLAIR image (Fig. 6.11.3) in a different patient demonstrates multiple foci of increased signal in the cortex and subcortical white matter consistent with tubers. Multiple low-signal subependymal nodules are also present. Axial unenhanced CT (Fig. 6.11.4) in another patient shows subependymal calcifications (arrowheads) and multiple high- and low-attenuating cortical and subcortical tubers (arrows).
DIAGNOSIS: Tuberous sclerosis with subependymal giant cell astrocytoma
DISCUSSION: Tuberous sclerosis complex (TSC) is the second most common of the neurocutaneous syndromes and is characterized by the formation of hamartomatous lesions in multiple organs. The genes responsible for TSC are tumor suppressor genes TSC1 (9q34) that encodes the protein hamartin and TSC2 (16p13) that encodes the protein tuberin. Most cases (66%) of TSC result from a spontaneous mutation, and the remainder are inherited in an autosomal dominant fashion. Although there is complete penetrance, TSC exhibits a high degree of phenotypic variability. Many patients have obvious signs at birth, whereas others remain undiagnosed for many years. In addition, approximately 20% of TSC patients do not have either of the TSC1 or TSC2 mutations. The classic clinical triad of adenoma sebaceum, seizures, and mental retardation is present only in a minority of patients with TSC.
The CNS lesions include astrocytic hamartomas of the retina, cortical tubers, and subependymal nodular hamartomas. A single cortical/subcortical lesion as seen in focal cortical dysplasia of Taylor (balloon type) is histologically the same as TSC. Approximately 15% of patients develop subependymal giant-cell astrocytomas (GCA), which are characterized as grade 1 neoplasms by the WHO. The GCAs virtually always arise near the foramen of Monro and can cause obstructive hydrocephalus. GCAs may also undergo malignant degeneration. By CT, GCA appears as an enhancing soft-tissue mass with variable calcification. MR imaging shows hypointensity to isointensity on T1-weighted images, hyperintensity on T2-weighted images, and uniform gadolinium enhancement. Even subependymal hamartomas frequently enhance on MRI, and interval increase in size is considered the only reliable sign of GCA development (18,21,22).
Lesions associated with TSC outside of the CNS include renal angiomyolipomas (AML), lymphangiomyomatosis (LAM) of the lung, and cardiac rhabdomyomas.
Aunt Minnie’s Pearls
The CNS lesions of TSC include cortical tubers, subependymal and retinal hamartomas, and GCAs. Interval increase in size of subependymal hamartomas near the foramen of Monro on MRI indicates development of GCAs.
Renal AMLs, pulmonary LAM, and cardiac rhabdomyomas are associated with TSC.
Case 6.12
HISTORY: A 32-year-old man with ataxia, nausea, and vomiting
 FIGURE 6.12.1 |
 FIGURE 6.12.2 |
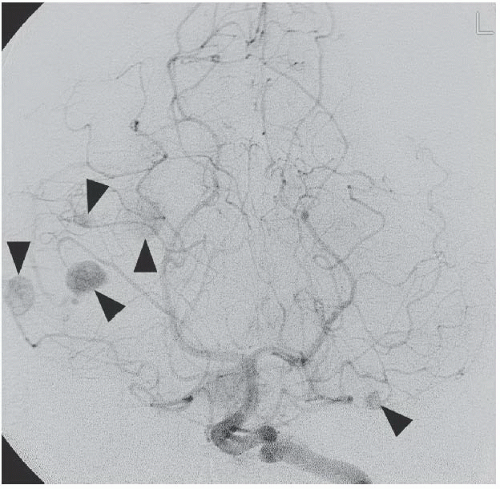 FIGURE 6.12.3 |
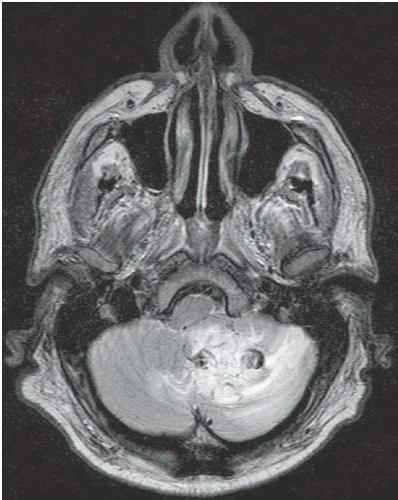 FIGURE 6.12.4 |
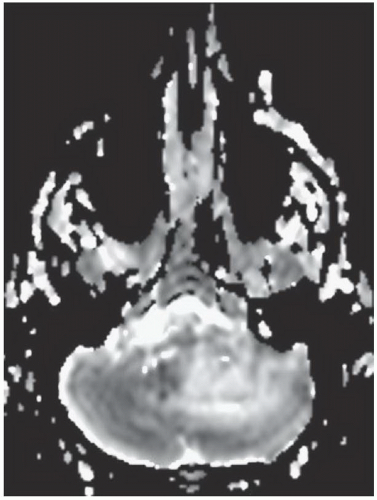 FIGURE 6.12.5 |
FINDINGS: Axial, T1-weighted post-contrast MR image (Fig. 6.12.1) demonstrates two enhancing masses in the right cerebellar hemisphere, the larger one with a cystic appearance containing a mural nodule (arrowhead). The other lesion is more laterally and anteriorly located (arrow). Sagittal post-contrast T1-weighted image through the cervical spine (Fig. 6.12.2) shows an additional enhancing lesion along the dorsal spinal cord surface. AP digital subtraction angiogram from a left vertebral injection in the same patient (Fig. 6.12.3) demonstrates multiple cerebellar hypervascular masses (arrowheads




Related posts:



Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
