The concept of segmental vascular syndromes with different, seemingly unrelated, diseases is based on the embryology of the neural crest and the mesoderm migration of cells that share the same metameric origin. Migrating patterns of these cells link the brain, the cranial bones, and the face on the same side. A somatic mutation developing in the region of the neural crest or the adjacent cephalic mesoderm before migration can, therefore, be postulated to produce arterial or venous metameric syndromes, including PHACES, CAMS, Cobb syndrome, and Sturge-Weber syndrome. Although these diseases may be rare, their relationships among each other and their postulated linkage with the development of the neural crest and the cephalic mesoderm may shed light on the complex pathology and etiology of various cerebral vascular disorders.
In 1879, Sturge reported a case that had an extensive port wine stain of the right face and head with a right-sided buphthalmus and a seizure attack. He also found a vascular malformation involving the ipsilateral brain. The atrophy of the affected cerebral hemisphere was suggested first by Weber in 1922 by using radiograph examinations of the skull. In 1936, Bergstrand and colleagues coined the currently accepted eponym Sturge-Weber disease and illustrated its pathologic characteristics, clinical manifestations, and surgical indications. Similarly, an association between arteriovenous malformations (AVMs) of the face, retina, and brain was recognized first by Bonnet, Dechaume, and Blanc , in Lyon, France, who reported two cases in 1937. Six years later, Wyburn-Mason reviewed all similar cases previously described and, in a detailed study, added nine more examples. The association of retinal, facial, and cerebral vascular malformations became known as Bonnet-Dechaume-Blanc syndrome in France and continental Europe, and as Wyburn-Mason syndrome in the English literature. Because the degree of expression of the syndromes’ components varies, both clinically and morphologically, some confusion existed regarding the use of the two names. The term Bonnet-Dechaume-Blanc was sometimes preferred for the more extreme end of the disease spectrum, which includes high-flow maxillofacial AVMs. However, careful reading of the original articles confirms that both descriptions were referring to the same condition; thus, both eponyms can be used interchangeably.
The association between AVMs of the spinal cord and the skin was named after Stanley Cobb, a resident under Harvey Cushing, who in 1915 described the case of an 8-year-old boy who presented with acute paraplegia and had a skin discoloration of the same metamere, identified as a nevus. On surgery, a large spinal AVM was noticed.
Lastly, the association of cervicofacial hemangiomas with vascular and nonvascular intracranial malformations was recognized first by Pascual-Castroviejo who, in 1978, proposed the name “cutaneous hemangioma-vascular complex syndrome.” In 1996, Frieden and colleagues proposed the acronym PHACE for this neurocutaneous syndrome; this acronym is derived from the spectrum of clinical findings that can be displayed in affected patients, (ie, P osterior cranial fossa malformations, H emangiomas, A rterial anomalies, C oarctation of the aorta and cardiac defects, and abnormalities of the E ye).
All the previously mentioned syndromes have been classified as neurocutaneous syndromes or phakomatoses. Within the past few years, however, the classic view of these phakomatoses has been revisited, and they no longer can be discussed without taking into consideration the recent genetic or biologic contributions that have suggested the neural crest and disorders in cephalic migration as a common link between the various components of many of these syndromes.
In this article, the authors first describe the concept of cephalic migration and then, based on this concept (ie, using a metameric approach), try to elucidate the previously mentioned syndromes and their common link.
General concepts: neural crest, cephalic migration, and the arterial-veno-lymphatic tree
The neural crest and neural plate share a common lineage. Cells of the lateral border of the developing neural plate (ie, the neural folds), under the inductive influence of the adjacent epithelium and possibly the mesoderm, develop into crest cells; hence, their common metameric origin with the cells of the hindbrain . Moreover, it appears that all neural plate cells can become crest cells given the appropriate signals, and vice versa, and even epithelial cells can, in vitro, contribute to the neural crest lineage. They also share a metameric relationship with the adjacent cephalic mesoderm. The neural crest gives rise to a wide range of cell types, including skin, connective tissue, the skeleton of the craniofacial region, forebrain meninges, and the tunica media of the blood vessels of the face and forebrain (the endothelial cells here and elsewhere are derived from mesoderm). In recent years, the Hox genes–controlled segmentation of the rhombencephalon into rhombomeres, and of the forebrain into prosomeres has been substantiated in birds, mice, and other animals, and extrapolated to humans . After Le Douarin’s introduction of the quail-chick chimera system in 1969, which allows labelling of cells in avian embryos and then following their migration to their definitive sites, studies have revealed the metameric nature of brain and craniofacial structures deriving mainly from the neural crest and plate . The initial process of vessel formation in the embryo, known as vasculogenesis, involves differentiation and sprouting of mesoderm-derived endothelial cells to form the primitive capillary network, which is then extended and remodeled by angiogenesis . These primary capillary vessels progressively become ensheathed by differentiating smooth muscle cells. Although head and neck endothelial cells, as elsewhere, derive from mesoderm, the tunica media of these vessels differentiates from neural crest cells , which stream into the developing head and pharyngeal arches. Hox gene–encoded positional information in the crest cells is known to be involved in patterning of the pharyngeal arches, and is most likely involved also in determining the neural crest cell distribution among the arch-derived arteries. The work of Couly and colleagues has shown further that the neural crest and mesodermal cells originating from a given transverse (metameric) level of the embryo finally occupy the same territory in the head, and that these embryonic tissues are regionalized in various areas devoted to providing blood vessels to specific regions of the face and brain.
A somatic mutation developing in the region of the neural crest or adjacent cephalic mesoderm before migration could, therefore, be expected to produce malformations with a segmental distribution. In fact, in avian experiments, fate maps of the cells occupying specific regions of the neural plate, crest, and cephalic mesoderm revealed striking similarities to the distribution of lesions encountered, for example, in the Wyburn-Mason syndrome ( Fig. 1 ) .
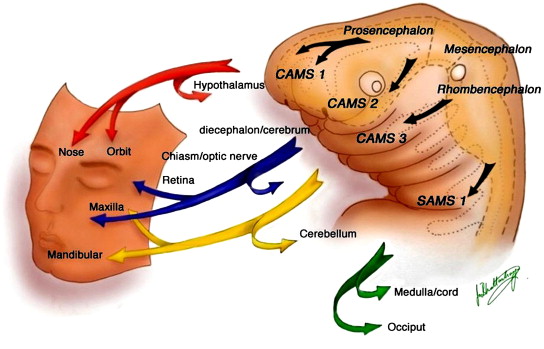
In addition to their similarities in segmental distribution pattern and their link to the concept of neural crest development, metameric origins of cells, and cephalic migration, these segmental vascular syndromes share features such as their evolutive nature over time and their potential incomplete expression. The former may lead to a progressive enlargement of some of the lesions, revealing potential angiogenic activity, or to a progressive expression in time. The latter demonstrates that all features of a specific syndrome may not be found at clinical presentation, because of differences in vulnerability, the specificity of the triggering event, or incomplete expression, as described earlier. Despite these similarities, the main difference between the herewith described segmental vascular syndromes (namely, cerebrofacial venous metameric syndromes [CVMS] , cerebrofacial or spinal arteriovenous metameric syndromes [CAMS or SAMS] , and PHACE syndrome [PHACES] ) is the target of the lesion found on different regions of the arterio-venous tree: on the venolymphatic end in CVMS, on the capillary-venous side in CAMS and SAMS, and on the arterial-capillary end in PHACES. Although the timing of the triggering event to produce a metameric syndrome as described in this article is the same (fourth to fifth embryonic week), the target differs. Arteries and veins are not identified by flow characteristics, but reveal their differences early by molecular properties. A specific trigger is therefore likely to be selective enough to differentiate between the more arterial and the more venous target, which is why, for example, port wine stains are not associated with intracranial AVMs. A striking difference between CVMS and CAMS on the one side and PHACES on the other is that, although all three syndromes share the migrating pattern of the neural crest and cephalic mesoderm along their three main paths, the clinical manifestations associated with PHACES (ie, disorders at the level of the sternum, the aorta, the internal carotid and intracranial arteries, the eye, and the posterior fossa) suggest a longitudinal dysfunction of the cephalic neural crest and are different from the axial, segmentally arranged vascular syndromes found in CAMS and CVMS, with lesions found within a single metamere (see Fig. 1 ). The association of hemangiomas in the cerebello-pontine angle with ipsilateral agenesis of the internal carotid, as described later, as potential manifestations of PHACES points to the link that exists in this syndrome between the third aortic arch, the third branchial arch, and the romboencephalic pattern of migration, also demonstrated in CAMS and CVMS. Finally, although CAMS and CVMS are well differentiated cranially (presumably because of their origin form the neural crest), sometimes they are not well delineated from each other at the spinal level (where the neural crest does not contribute directly to vasculogenesis). Therefore, the venolymphatic Klippel-Trenaunay syndrome may be associated with spinal cord arterial aneurysms (AA) and AVMs, whereas port wine stains (as a typical venous malformation) can be associated with typical SAMS (as a more arterial disease).
Cerebrofacial and spinal arteriovenous metameric syndromes
The association of AVMs of the brain, the orbit (retinal or retrobulbar lesions), and the maxillofacial region was named originally after Bonnet-Dechaume-Blanc and Wyburn-Mason. Because of the previously mentioned metameric concept of neural crest development, the authors have proposed a rational classification reflecting the putative underlying disorder, and have coined the acronym CAMS. Depending on the involved structures, several CAMS can be differentiated: CAMS 1 as a midline prosencephalic (olfactory) group with involvement of the hypothalamus, corpus callosum, hypophysis, and nose ( Fig. 2 ); CAMS 2 as a lateral prosencephalic (optic) group with involvement of the optic nerve, retina, parieto-temporal-occipital lobes, thalamus, and maxilla ( Fig. 3 ); and CAMS 3 as a rhombencephalic (otic) group with involvement of the cerebellum, pons, petrous bone, and mandible ( Fig. 4 ). CAMS 3 is located in a strategic position on the crossroad between the complex cephalic segmental arrangements and the relatively simplified spinal metamers, and therefore it may bear transitional characteristics . SAMS, previously referred to as Cobb’s syndrome or spinal AVM of the juvenile type, likewise affects the whole myelomere; therefore, affected patients typically present with multiple shunts of the spinal cord, nerve root, and bone, and paraspinal, subcutaneous, and skin tissues that share the same myelomere ( Fig. 5 ) . SAMS can be named from 1 to 31, depending on the affected myelomere . A more extensive insult leads to overlapping territories, producing a complete prosencephalic phenotype (CAMS 1 + 2) or bilateral involvement. The insult producing the underlying lesion would have to develop before the migration occurs and, thus, before the fourth week of development. The disease spectrum may be incomplete either because some cells are spared or because they have not been triggered to reveal the disease , leading to cases without retinal involvement , cerebral involvement, or facial involvement . Retinal AVM is often the earliest manifestation of a CAMS and in some cases follow-up has shown secondary expression of the full syndrome. In one case, the full extent of the spectrum was revealed over a 28-year period . Anticipation of the other localizations can be discussed when isolated retinal, hypothalamic, or optic nerve AVM is diagnosed.
Retinal Arteriovenous Malformations and Arteriovenous Malformations Along the Optic Nerve and Chiasm
The most common presenting symptom of retinal AVMs and AVMs along the optic nerve and chiasm is visual deterioration (reduced acuity or field) because retinal AVMs are present in most CAMS patients. These symptoms occur before the age of 18 in more than 50% of cases. Usually, retinal AVMs or arteriovenous communications of the retina (AVCR) are considered to be stable retinal lesions, despite progression of the coexisting intracranial AVMs . Loss of vision may occur because of intraretinal macular hemorrhage, central and peripheral retinal vein occlusion, or vitreous hemorrhage; a gradual reduction of vision may be caused by neovascular glaucoma in association with changes in the retinal AVMs and retinal and choroidal ischemia . The retinal AVM or AVCR is called a racemose hemangioma in the ophthalmologic literature (although racemose hemangioma is probably a misnomer because it does not exhibit proliferative behavior). Sometimes it is detected several years before further neurologic symptoms lead to more detailed investigations (CT, MR imaging) confirming the presence of a retinal AVM, but also revealing an associated brain AVM.
Optic nerve and chiasmatic AVMs are also hallmark findings in CAMS. They often appear to be clinically silent, eventually resulting in a slowly progressive, functional deficit. The presenting symptoms include decreased visual acuity and field defects, or blindness from optic nerve atrophy and progressive dysfunction of the optic pathways.
Exophthalmos is a rare presenting symptom in CAMS, with optic nerve AVMs and intraorbital congestion resulting in mass effect. Exophthalmos can also result from an enlarged ophthalmic vein, which may drain normal brain tissue, or from cerebral AVMs related to increased intracranial venous pressure caused by venous occlusion of draining intracranial veins.
Treatment (a combined surgical and endovascular approach) of these orbital lesions remains a challenge because of the complex anatomy and hemorrhagic characteristics of the malformation .
Cerebral Arteriovenous Malformations
Considered metameric lesions, intracranial AVMs in CAMS are one of the most common findings in such patients. Cerebral AVMs in CAMS may involve in continuity the corpus callosum, the olfactory region, and the hypothalamus (CAMS 1); the optic chiasm, the thalamus, and the cortex around the calcarine fissure (CAMS 2); or the cerebellum (CAMS 3). Infrequently, they present as multiple scattered lesions in the same segmental distribution. Considering the angioarchitecture of cerebral AVMs in CAMS, certain findings differ from “classic” AVMs: the AVM nidus in CAMS is a cluster of small vessels with intervening normal brain tissue, some degree of angiogenesis, and a small shunting volume . Transdural arterial supply can be present. Progressive enlargement of these cerebral AVMs is one of the special observations in CAMS, suggesting that AVMs in CAMS are not static processes within the segment that carry the embryonic defect. Multifocality is another typical aspect of CAMS-associated AVMs. Despite the common occurrence of cerebral AVMs in CAMS, they are usually clinically silent or asymptomatic at the time of discovery. They rarely present with acute neurologic symptoms caused by intracerebral or subarachnoid hemorrhage ; rather, they reveal with progressive neurologic deterioration without evidence of intracranial bleeding, most likely owing to a progression in size. About 25% of patients who have CAMS-associated AVMs bleed during the course of their disease .
Therapeutic management of cerebral AVMs is particularly challenging. The authors suggest targeted embolization in an attempt to exclude weak angioarchitectural aspects or to reduce the arteriovenous (AV) shunt in the least eloquent areas in symptomatic patients who are significantly affected clinically. At present, brain AVMs are not considered curable because of their size, location, and natural history .
Facial Arteriovenous Malformations, Nasal Arteriovenous Malformations, and Mandibular Arteriovenous Malformations
Nasal AVMs (CAMS 1), maxillofacial AVMs (CAMS 2), and mandibular AVMs (CAMS 3) are present in about one third of patients who have CAMS. The presence of the facial vascular lesion can be difficult to recognize or may be clinically silent, sometimes representing a small stable red spot or angioma since infancy or early childhood. Then, an unknown trigger occurs that promotes growth of the lesion, with revealing symptoms such as bleeding of the gums or mass effect, resulting in facial asymmetry, often during adolescence. Epistaxis may be encountered in high-flow maxillofacial AVMs. The angioarchitecture of facial or mandibular AVMs in CAMS looks similar to sporadic cases. Usually, they present as an arteriovenous fistula or nidus-type AVM with intranidal fistulous compartments . Large, proximal arterial aneurysms can be present, despite relative slow flow or small-sized facial or mandibular AVMs. Because sporadic, isolated maxillar or mandibular AVMs are not associated with external carotid artery (ECA) aneurysms regardless of their flow, facial AVMs in CAMS patients are different from their sporadic counterparts in terms of angioarchitecture and natural history. Maxillar or mandibular AVMs with intraosseous venous lakes or pouches are also at risk of gum bleeding and severe hemorrhages after tooth extraction . The presence of atypical aneurysms on the ECA in CAMS patients points toward the angiogenic potential of this syndrome.
Cerebrofacial and spinal arteriovenous metameric syndromes
The association of AVMs of the brain, the orbit (retinal or retrobulbar lesions), and the maxillofacial region was named originally after Bonnet-Dechaume-Blanc and Wyburn-Mason. Because of the previously mentioned metameric concept of neural crest development, the authors have proposed a rational classification reflecting the putative underlying disorder, and have coined the acronym CAMS. Depending on the involved structures, several CAMS can be differentiated: CAMS 1 as a midline prosencephalic (olfactory) group with involvement of the hypothalamus, corpus callosum, hypophysis, and nose ( Fig. 2 ); CAMS 2 as a lateral prosencephalic (optic) group with involvement of the optic nerve, retina, parieto-temporal-occipital lobes, thalamus, and maxilla ( Fig. 3 ); and CAMS 3 as a rhombencephalic (otic) group with involvement of the cerebellum, pons, petrous bone, and mandible ( Fig. 4 ). CAMS 3 is located in a strategic position on the crossroad between the complex cephalic segmental arrangements and the relatively simplified spinal metamers, and therefore it may bear transitional characteristics . SAMS, previously referred to as Cobb’s syndrome or spinal AVM of the juvenile type, likewise affects the whole myelomere; therefore, affected patients typically present with multiple shunts of the spinal cord, nerve root, and bone, and paraspinal, subcutaneous, and skin tissues that share the same myelomere ( Fig. 5 ) . SAMS can be named from 1 to 31, depending on the affected myelomere . A more extensive insult leads to overlapping territories, producing a complete prosencephalic phenotype (CAMS 1 + 2) or bilateral involvement. The insult producing the underlying lesion would have to develop before the migration occurs and, thus, before the fourth week of development. The disease spectrum may be incomplete either because some cells are spared or because they have not been triggered to reveal the disease , leading to cases without retinal involvement , cerebral involvement, or facial involvement . Retinal AVM is often the earliest manifestation of a CAMS and in some cases follow-up has shown secondary expression of the full syndrome. In one case, the full extent of the spectrum was revealed over a 28-year period . Anticipation of the other localizations can be discussed when isolated retinal, hypothalamic, or optic nerve AVM is diagnosed.
Related posts:
 Endovascular Treatment of Hemangiomas
Endovascular Treatment of Hemangiomas
 Diagnosis and Endovascular Treatment of Pediatric Spinal Arteriovenous Shunts
Diagnosis and Endovascular Treatment of Pediatric Spinal Arteriovenous Shunts
 Pediatric Neuroanesthesia
Cerebral Sinovenous Thrombosis in Children
Pediatric Neuroanesthesia
Cerebral Sinovenous Thrombosis in Children
 Overt and Incomplete (Silent) Cerebral Infarction in Sickle Cell Anemia: Diagnosis and Management
Current Endovascular Management of Maxillofacial Vascular Malformations
Overt and Incomplete (Silent) Cerebral Infarction in Sickle Cell Anemia: Diagnosis and Management
Current Endovascular Management of Maxillofacial Vascular Malformations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


