Neurofibrillary pathology in Alzheimer’s disease consists of paired helical filaments comprising tau protein. This pathology is correlated with dementia, but can appear in the first two decades of life. Extracellular amyloid β-protein arises through proteolytic processing of a transmembrane precursor, which involves the action of several enzymes. Mutations in the genes for the precursor and presenilin proteins accelerate the deposition of Aβ. Tau mutations cause other tauopathies in the absence of amyloid deposition, indicating that amyloid deposition is not a prerequisite for dementia. An improved understanding of Alzheimer’s disease awaits to be obtained by molecular imaging of these pathologies.
- •
The pathologic hallmarks of Alzheimer’s disease are the presence of both neurofibrillary tangles and senile plaques, first described in a patient with presenile dementia by Alois Alzheimer.
- •
Neurofibrillary pathology consists of intraneuronal fibrils present in tangles, and in neurites found both throughout the neuropil and in neuritic senile plaques.
- •
Neurofibrillary pathology in Alzheimer’s disease consists of paired helical filaments comprising tau protein.
- •
Amyloid pathology occurs as the deposition of amyloid-β (Aβ) protein within senile plaques and in the form of diffuse deposits throughout the neuropil. In addition, it can be found as deposits around blood vessels.
- •
Aβ deposits derive from the processing of a transmembrane-spanning Aβ protein precursor (APP) to release an extracellular Aβ peptide of 40 to 43 amino acids in length; larger peptides form more insoluble Aβ deposits.
- •
Mutations in APP and the presenilin proteins 1 and 2 cause familial Alzheimer’s disease and result in increased deposition of insoluble Aβ. Presenilin 1 possesses the catalytic sites of a tetrameric γ-secretase complex that cleaves APP, with the aid of β-secretase, to Aβ peptides.
- •
Tau mutations cause tauopathy in conditions other than Alzheimer’s disease in the absence of amyloid pathology. These conditions include frontotemporal dementia syndromes, and indicate that amyloid is not essential to cause dementia.
- •
Tau pathology in Alzheimer’s disease precedes amyloid pathology by 2 decades.
- •
Molecular imaging of amyloid in Alzheimer’s disease using Pittsburgh B compound and the future development of selective tau ligands indicate that knowledge of the pathogenesis of Alzheimer’s disease will be greatly improved by direct visualization of the pathology in humans.
The German psychiatrist Alois Alzheimer presented the original description for a 55-year-old patient who had suffered with presenile dementia and who had the unique combination of both senile plaques and neurofibrillary degeneration in her brain. Alzheimer was prompted to consider this case as representing a specific disease process in which the neurofibrillary pathology made it particularly distinctive. In just 2 pages, Alzheimer had described the pathologic basis for the disease to be given his name. By 2010, more than 9000 articles related to Alzheimer’s disease were published in a single year.
The pathologies that Alzheimer observed at autopsy can now be examined during life by molecular imaging techniques that allow an even greater insight into the pathogenesis of the disease and the means to assess therapeutic efficacy. This article focuses on the molecular pathology found in Alzheimer’s disease, namely that of tau protein and amyloid-β (Aβ) protein ( Fig. 1 ).
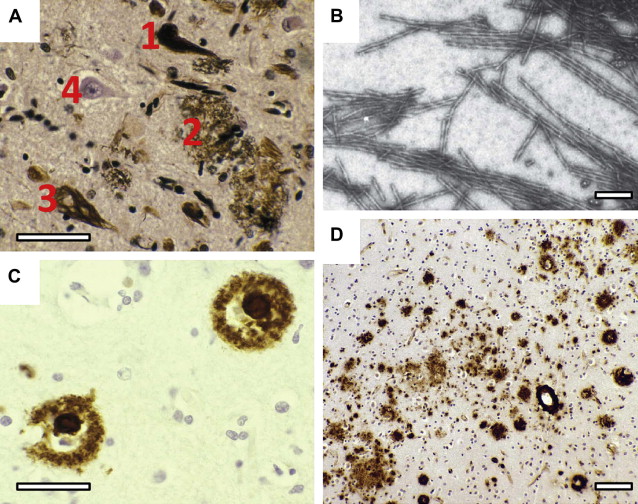
Tau protein pathology in Alzheimer’s disease
Normal Tau Protein
Tau proteins are a family of microtubule-associated proteins. Tau proteins are predominantly expressed in neurons, where they play an important role in the assembly and stabilization of tubulin monomers into microtubules that constitute the neuronal cytoskeletal network. Microtubules are essential in morphogenesis, cell division, and intracellular trafficking of organelles. At physiologic concentrations tau proteins stabilize microtubules as tracks for intracellular transport, but in excess they interfere with transport down the axon. Tau also plays a role in signal transduction through its interaction with phospholipase C-γ, interacts with actin and the plasma membrane, is involved in anchoring of protein kinases and phosphatases, and is important in neurite outgrowth.
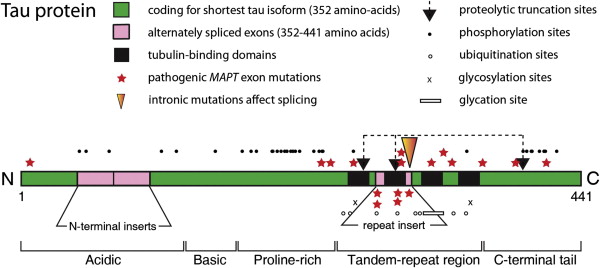
Human tau protein in the central nervous system exists in 6 isoforms, ranging from 352 to 441 amino acids in length and derived by alternative mRNA splicing from a single gene ( MAPT ) located on chromosome 17. The domains of tau protein are depicted in Fig. 2 . There are 2 inserts in the acidic N-terminal domain encoded by exons 2 and 3. Each isoform contains either 3 or 4 tandem repeats of 31 or 32 amino acids located in the C-terminal portion of the molecule. The additional repeat is encoded by exon 10. These repeats are both rich in basic amino acids and capable of binding to an acidic domain of tubulin. Similar microtubule-binding repeats are found in the carboxy-terminal domain of high molecular weight microtubule-associated proteins (MAPs), such as MAP2. The different MAPs serve specific functions; tau is predominantly axonal in location, whereas MAP2 is restricted to the somatodendritic part of the neuron.
Neurofibrillary tangles (NFTs) are intraneuronal inclusions that form in a few susceptible types of neurons. NFTs are composed of aberrant tau protein polymers in the form of paired helical filaments (PHFs) (see Fig. 1 B). There is considerable evidence in support of Alzheimer’s view on the importance of neurofibrillary tangles in dementia. The neurofibrillary tau pathology is closely linked to intellectual status antemortem, and deposition of diffuse amyloid can be extensive in intellectually normal elderly cases. By contrast, there are no reports of preservation of normal cognitive functioning in the presence of numerous neocortical tangles. The density of neuritic plaques, rather than amyloid deposits, tends to correlate better both with the biochemical measure of PHF-tau and with cognitive dysfunction. Further studies also confirm that Aβ load does not predict neuronal loss, synapse loss, or dementia. Furthermore, there is considerable overlap in the levels of Aβ deposition found in Alzheimer’s disease and nondemented patients, which is not the case with PHF accumulation. The PHF-tau content in Alzheimer’s disease tissue exceeds that found in control tissue by 19-fold, and the difference is even greater for brain regions such as temporal cortex. The accumulation of PHFs is accompanied by a corresponding loss of normal soluble tau, in excess of the loss due to normal aging. By contrast, the levels of Aβ in nearly half of the cases of Alzheimer’s disease are found to overlap the levels found in controls, regardless of APOE genotype. These findings argue against the hypothesis that Aβ deposition induces a direct toxic effect on neurons in the human brain, and rather poses the question as to exactly how tau and amyloid pathologies are connected.
Tau pathology is closely linked with synaptic loss, and Terry and colleagues first reported that loss of synapses provided a better correlate of cognitive deficit than neurofibrillary tangles, a finding supported by others. The tauopathy of Alzheimer’s disease follows a characteristic pattern of spread of tau aggregation pathology, initially described by Braak and Braak ( Fig. 3 ). NFT pathology is most severe when it begins in the hippocampus and entorhinal cortex and, with progression, spreads to temporal, parietal, and frontal cortices. The process of tau aggregation begins in the neocortical regions well before NFTs appear. Tau aggregation, in the form of proteolytically stable PHF-tau, is detected from Braak stage 2 onwards, whereas tangles that can be visualized by conventional microscopy do not appear until Braak stage 4 in the neocortex.
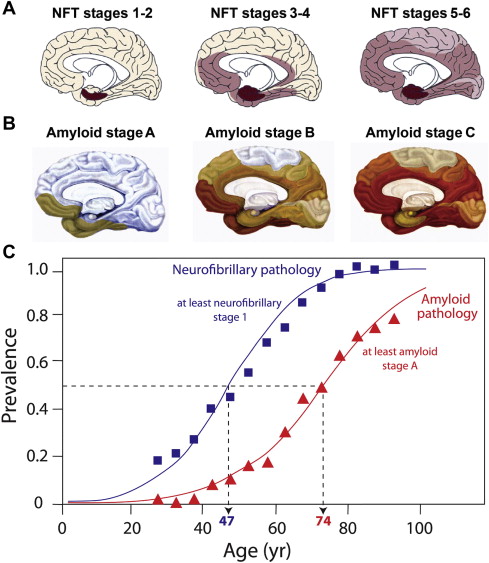
Duyckaerts recently compared the temporal sequence of the appearance of tau and amyloid pathology from previously published Braak data to show that tau pathology in the medial temporal lobe precedes Aβ accumulation by more than 2 decades (see Fig. 3 ). Furthermore, tau pathology is observed in a high proportion of children in the absence of Aβ accumulation.
Mutations in the MAPT Gene that Cause Dementia
Mutations in the MAPT gene that cause neurodegenerative tauopathies other than Alzheimer’s disease, namely progressive supranuclear palsy, argyrophilic grain disease, and a variety of frontotemporal disease syndromes, have been identified. At the ultrastructural level, the tau filaments in these patients consist mainly of straight or twisted-ribbon filaments rather than the PHFs typical of Alzheimer’s disease. Although mutations in the MAPT gene have not been found in Alzheimer’s disease, the discovery that such mutations can cause dementia in the absence of β-amyloid pathology asserts that tau pathology need not be a secondary consequence of amyloid deposition.
There are 2 main classes of tau mutations: those that affect alternative splicing of the MAPT gene and those that directly affect the function of the protein. Mostly they cluster in the vicinity of the tandem-repeat domain of tau, as shown in Fig. 2 . Mutations in the 5′-splice site of exon 10 destabilize the intron 10 stem-loop structure, which increases production of the 4-repeat isoforms. Recombinant tau proteins carrying missense mutations or the deletion mutation (K280Δ) have a decreased ability to promote microtubule assembly, which is more marked for 3-repeat than for 4-repeat isoforms. A decrease in the binding of tau to microtubules could destabilize them over time and disrupt axonal transport. The tau mutations could also lead to increases in free cytosolic tau and an increased propensity to form insoluble tau aggregates. Of the mutations tested, the P301L and K280Δ mutations in exon 10 have the greatest effect on microtubule assembly. The P301L mutation has the greatest potential for fibril formation, and spherical structures are obtained by incubation of the mutant protein in the absence of heparin. Furthermore, filaments are produced in the brains of mice that are transgenic for this mutant form of tau protein. The mutations N279K and S305N, on the other hand, do not show any decreased ability to promote microtubule assembly. The latter mutations increase the splicing-in of exon 10. It is possible that subtle changes in the ratio between 4-repeat and 3-repeat tau isoforms may be sufficient to cause neurodegeneration over a prolonged period. Greater distances between the microtubules and fewer microtubules per process were observed with the V337M mutant isoform. The varied ways by which the mutations affect the processing and function of tau are probably responsible for the phenotypic heterogeneity observed in patients with FTDP-17 (frontotemporal dementia with parkinsonism–17).
Posttranslational Modifications of Tau Protein
The distinctive feature of Alzheimer’s disease is the substantial redistribution of tau protein from its normal axonal location into somatodendritic PHFs, the mechanism whereby this occurs being poorly understood. Tau protein is subject to several posttranslational modifications that may affect this distribution, including ubiquitination, phosphorylation, glycosylation, nonenzymatic glycation, and oxidation. Sites where these changes occur are depicted schematically in Fig. 2 . Further changes that are observed in Alzheimer’s disease are the proteolytic truncation of tau and its aggregation into oligomers and PHFs.
Expression of the different tau isoforms is developmentally regulated, and the extent of phosphorylation and glycosylation affects the function of the protein. A combination of kinase and phosphatase activity contributes to the extent of phosphorylation of tau, both in normal situations and in Alzheimer’s disease. Thus, tau from the neonate is phosphorylated at more sites than is tau from adult brain, and tau phosphorylation in developing brain is more dynamic than in adult brain. Phosphorylated tau is unable to bind microtubules, although the phosphorylation of tau has been found to be inhibitory for the aggregation of tau in vitro.
O -Linked N -acetyl glucosamination is a dynamic and abundant posttranslational modification that can operate reciprocally with Ser-/Thr- phosphorylation. Phosphorylation of Ser-/Thr- residues in PHF-tau would decrease the occurrence of O -GlcNAc in PHFs. Modification of PHF-tau, but not normal tau, by N -linked glycosylation has been reported. By contrast, N -linked glycosylation usually occurs cotranslationally and is considered a less dynamic modification. Deglycosylation of PHFs, however, converts them into straight filaments, suggesting that N -linked glycosylation may contribute to the maintenance of the PHF structure.
It is likely that ubiquitination, nonenzymatic glycation, and oxidation are late events that arise in older neurons as levels of oxidative stress increase with aging. The first 2 of these changes are observed in the vicinity of the tandem-repeat region of tau, where the insoluble tangles are probably subject to futile attempts by the neuron to remove proteolytically resistant PHF-tau. By contrast, the role of phosphorylation at sites outside the repeat domain is more dynamic, affecting the numerous surface-exposed substrates for the abundant cytoplasmic kinases and phosphatases.
Aggregation of Tau Protein in Alzheimer’s Disease
Normal tau protein possesses little physical structure: α-helical, β-sheet, and β-turns are absent. Nevertheless, tau aggregation through the repeat domain confers proteolytic stability on a short segment of the molecule that corresponds closely to that isolated from the PHF core. This fragment has the intrinsic capacity to propagate tau capture. Although extracellular matrix proteins (heparin) and acidic proteins (RNA) can promote tau aggregation in vitro, facilitation of tau aggregation may be attributable to a general property of macromolecular substrates that bind tau nonspecifically. The substrates that initiate tau aggregation in vivo remain to be identified. It is possible that stochastic conformational changes to tau molecules occur with aging to cause sporadic Alzheimer’s disease. Alternatively, conformational changes might be induced through interaction with other macromolecules, and tau aggregation may be seeded by cross-linked dimers or preformed tau aggregates extracted from the brain.
Several neurodegenerative diseases in which there is aberrant protein aggregation are characterized by protein misfolding. These disorders have been given terms conformational diseases or prionoses. The deposits consist of highly ordered fibrils having a cross-β-pleated structure. Despite the absence of homology in the primary structure of various precursor proteins, the amyloid structure is common for many different amyloid disorders.
Full-length tau does not aggregate in physiologic conditions, and this does not change when the protein has been hyperphosphorylated in vitro. Tau can be made to assemble into filaments in nonphysiologic buffers and at high protein concentrations, or when tau aggregation is facilitated by coincubation of tau protein with sulfated glycosaminoglycans. Typically tau concentrations of between 40 and 100 μM are required, whereas direct measurements in the human brain indicate that tau protein concentrations are unlikely to exceed 1 μM within pyramidal cells.
The tau protein present within the structural core of the PHF is a short fragment of nearly 100 amino acid residues in length, which derives from the repeat domain of tau. This core-tau unit of the PHF has the remarkable property that it is able to reproduce itself at the expense of normal tau. Normally, tau binds to tubulin monomers of the microtubule, as illustrated in Fig. 4 . In Alzheimer’s disease, however, tau accumulates as oligomers, which are subject to proteolysis, leaving a protease-resistant core. Such a core unit has the capacity to bind further tau molecules and initiate an autocatalytic process of tau aggregation. Subsequently, oligomers assemble into the PHFs, as shown schematically in the lower panel of Fig. 4 .
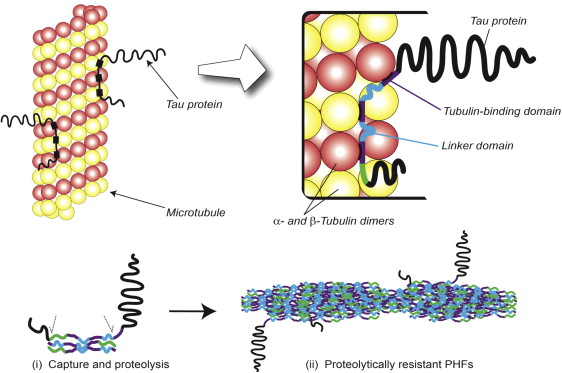
The capacity of tau protein pathology to propagate itself at the expense of normal tau has been confirmed in other studies. In an inducible tau transgenic model, tau pathology continued to progress even when expression of the tau transgene had been turned off. Aggregated fibrils of a truncated tau fragment (residues 243–375) can be taken up into cells containing full-length tau and passed on to neighboring cells to seed aggregation. Similarly, the transmission and spread of tau pathology from tau-mutant mice into normal mice was achieved whereby the latter mice were expressing human full-length tau protein. These studies validate the proposal made by Braak and Braak in 1991 that tau pathology spreads from the entorhinal cortex.
Once tau aggregation has been initiated, the only pathway available for clearance of the tau oligomers is the endosomal-lysosomal pathway, which becomes progressively congested with proteins from various sources, including Aβ. The capacity of neurons to clear proteolytically stable tau oligomers is therefore compromised, leading to uncontrolled progression of the tau aggregation process.
In summary, therefore, Alzheimer’s disease is characterized by altered neuronal metabolism of tau whereby the protein forms aggregates that then assemble into filaments having a defined paired helical structure. The appearance of oligomeric tau is correlated with cognitive deficit, and the progression of this process continues at an exponential rate, just as the prevalence of Alzheimer’s disease increases in an age-dependent manner.
β-Amyloid pathology and molecular genetics of Alzheimer’s disease
Mutations that cause Alzheimer’s disease and influence Aβ protein deposition have been identified in 3 genes: Aβ protein precursor ( APP ) and 2 presenilin genes, PSNL1 and PSNL2 . An updated list of these mutations can be found at http://www.alzforum.org . Further genetic risk factors, such as phosphatidylinositol-binding clathrin assembly protein (PICALM), are being identified through genome-wide association studies (GWAS).
The Alzheimer’s disease mutations of APP are either within or flanking the Aβ domain of APP, a domain that gives rise to the deposits of Aβ plaques throughout the cortex and, to a lesser extent, in the cerebellum. APP exists in at least 5 isoforms, but its physiologic role in the brain is not understood. In the numbering of the largest APP isoform (770 residues), the 42-residue Aβ domain corresponds to residues 672 to 713 encoded for by exons 16 and 17 ( Fig. 5 ). In one family, a Swedish mutation results in the replacement of 2 amino acids (K670N and M671L), whereas other point mutations causing Alzheimer’s disease include A692G, E693G, V715M, I716L, V717I, V717G, V717F, V717L, and L723P (see Fig. 5 ). Hereditary cerebral hemorrhage with amyloidosis—Dutch type, which exhibits severe congophilic angiopathy in the absence of plaques and tangles, is caused by the mutation E693Q.
Related posts:
 Spatial Distribution and Secular Trends in the Epidemiology of Alzheimer’s Disease
Spatial Distribution and Secular Trends in the Epidemiology of Alzheimer’s Disease
 Clinical and Research Diagnostic Criteria for Alzheimer’s Disease
Clinical and Research Diagnostic Criteria for Alzheimer’s Disease
 Frontotemporal Lobar Degeneration: New Understanding Brings New Approaches
Frontotemporal Lobar Degeneration: New Understanding Brings New Approaches
 Frontotemporal Lobar Degeneration: New Understanding Brings New Approaches
Frontotemporal Lobar Degeneration: New Understanding Brings New Approaches
 Molecular Neuroimaging in Alzheimer’s Disease
Molecular Neuroimaging in Alzheimer’s Disease
 The Clinical Value of Large Neuroimaging Data Sets in Alzheimer’s Disease
The Clinical Value of Large Neuroimaging Data Sets in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


