Fig. 1
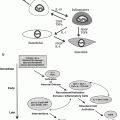
Simplified model of processes involved in the pathogenesis of radiation-induced lung injury. As noted in the diagram, each event has the potential to influence several other processes. Exposure to ionizing radiation initiates a cascade of cytokines and growth factors. Proinflammatory cytokines promote an influx of macrophages and inflammatory cells, which are stimulated to produce ROS, proinflammatory and profibrotic cytokines. ROS serve as redox regulators of transcription factors, which further stimulate induction and activation of cytokines and growth factors. In addition, vascular changes, as well as an increase in oxygen consumption by activated macrophages, contribute to the development and perpetuation of hypoxia and chronic oxidative stress, leading to the non-healing tissue response of chronic radiation injury. ACE = angiotensin converting enzyme; PA = plasminogen activator; PG = prostaglandins; Ang2 = angiotensin II; CAIX = carbonic anhydrase IX, HIF = hypoxia inducible factor; PDGF = platelet derived growth factor; IGF = insulin-like growth factors; bFGF = basic fibroblast growth factor. [Reproduced with permission from Fleckenstein et al. (2007a)]
Many of the molecular mediators of normal tissue injury are proteins, which can be measured both in tissue and in blood. The ability to quantify the expression of these proteins, in the normal and diseased state, has led to attempts to use them as predictors of risk of normal tissue injury after radiation therapy (Anscher et al. 1993, 1994; Vujaskovic et al. 1997; Chen et al. 2001; De Jaeger et al. 2004; Gridley et al. 2004; Novakova-Jiresova et al. 2004). Until recently, each protein had to be quantified individually using methods such as antibody-based enzyme-linked immunosorbent assays (ELISA) or bioluminescence assays, which are laborious and time consuming (Kong et al. 1998). Advances in bioassay technology now permit researchers to quantify multiple proteins simultaneously from the same sample in a rapid and reproducible manner (Jones et al. 2002). This technology will greatly enhance the ability to construct protein expression profiles for individual patients and determine whether these patterns of protein expression can improve our ability to predict risk of injury from radiation therapy (Hart et al. 2004). Along these lines, blood and tissue banks stocked with samples from patients irradiated for various malignancies will become invaluable resources for normal tissue injury research.
3 The Importance of Transforming Growth Factor ß1 in Radiation-Induced Injury
The most widely studied potential mediator of radiation-induced normal tissue injury is Transforming Growth Factor-ß1 (TGFß1). TGFß1 has multiple functions that are important in the development of excess fibrous tissue, one of the hallmarks of late radiation injury. TGFß1 is a chemoattractant for fibroblasts and also promotes differentiation of immature fibroblasts into myofibroblasts, which leads to increased production of collagen and extracellular matrix (Rodemann and Bamberg 1995, 2007). TGFß1 also decreases production of matrix-specific proteases and increases production of protease inhibitors, resulting in decrease collagen degradation, with a net result of increased fibrous tissue formation (Hakenjos et al. 2000; Martin et al. 2000). In addition to being autocrine stimulated, TGFß1 production is also stimulated by hypoxia, which further promotes collagen formation (Haroon et al. 2000; Moeller et al. 2004; Jackson et al. 2007).
Recent evidence confirms that TGFß1 is an important contributor to the pathogenesis of radiation-induced normal tissue injury. Rubin et al. (1992) reported that alveolar macrophages obtained from bronchial lavage specimens from irradiated rabbits demonstrated increased production and release of TGFß1 as compared to macrophages from normal lungs. These authors suggested that the fibroblast proliferation and extracellular matrix production found after irradiation are controlled by growth factors that are released from parenchymal cells following radiation exposure. Anscher et al. (1990) demonstrated that TGFß1 expression increased in a dose-dependent manner in the liver of rats following irradiation and that this increase in TGFß1 expression correlated with the extent of connective tissue production. Barcellos-Hoff (1995; Barcellos-Hoff and Dix 1996) has shown that free radicals produced during exposure to ionizing radiation can directly activate the latent form of TGFß1 which is sequestered in the extracellular matrix. It is likely that direct activation of TGFß1 from ionizing radiation contributes to the observed increase in TGFß1 levels 24 h post-radiation exposure (Fleckenstein et al. 2007b). Likewise, TGFß1 mRNA has also been shown to be elevated following radiation (Rube et al. 2000, 2004). Thus, radiation therapy can both increase local expression, as well as TGFß1 activation, resulting in increased fibrosis in irradiated tissues. As further evidence to support the role of TGFß1 in radiation injury, mice lacking Smad 3 (part of the TGFß1 signal transduction pathway) have been shown to be resistant to radiation-induced fibrosis (Flanders et al. 2002), suggesting that targeting the TGFß1 pathway might be a useful strategy to prevent radiation injury.
Indeed, several studies support the idea that reducing TGFß1 production may be one strategy to reduce normal tissue damage from radiation. Anscher and colleagues demonstrated that administration of a neutralizing TGFß1 antibody reduced radiation-induced macrophage accumulation, alveolar wall thickness, and TGFß1 activation in the lungs of rats (Anscher et al. 2006). More recently, a follow up study showed that a small molecule inhibitor of TGFß1 reduced breathing frequencies, lung fibrosis, inflammatory response, and TGFß1 activity in irradiated rats (Anscher et al. 2008). A similar conclusion was obtained by inhibiting integrin signaling in the lung, an important activator of the TGFß1 pathway (Munger et al. 1999). It was shown that integrin alpha(v) beta6-deficient mice were completely protected from radiation-induced fibrosis (Puthawala et al. 2008).
Local activation of TGFß1 in tissues may also be an important component in sustaining the process of abnormal wound healing long after the exposure to radiation has ended. For example, active TGFß1 both recruits and activates macrophages to secrete inflammatory and fibrogenic cytokines, including TGFß1 itself (Ashcroft 1999; Roberts et al. 2001). This auto-induction is important in maintaining levels of TGFß1 in wound healing. Following radiation, however, this process contributes to overproduction of collagen and inhibition of epithelial cell proliferation, increased local oxygen consumption by activated macrophages, and decreased oxygen delivery due to microvasculature injury creating a hypoxic environment (Li et al. 2001). These combined factors perpetuate normal tissue injury. In addition, sustained overproduction of TGFß1 may contribute not only to chronic fibrosis, but may also reduce the effectiveness of cancer therapies (Biswas et al. 2007) and contribute to the development of radiation-induced malignancy (see below).
4 Using Plasma TGFß1 Levels to Predict Injury Risk
Plasma TGFß1 levels recently has been used to try and identify patients at risk for the development of normal tissue injury after exposure to chemotherapy and/or radiotherapy. In patients who develop radiation-induced lung injury, Fu et al. (2001) found sustained elevations in plasma TGFß1 level for as long as 2 years after treatment. In contrast, patients who did not develop symptomatic lung injury did not exhibit sustained elevations in circulating plasma TGFß1. In another patient study, it was shown that TGFß1 blood plasma levels increased during the period of radiation treatment and also became significantly higher 4 weeks after radiotherapy (Kim et al. 2009). In addition, of several cytokines measured, only TGFß1 levels showed a correlation with the symptomatic occurrence of radiation pneumonitis (Kim et al. 2009). Recently, it was also shown that elevated levels of plasma TGFß1 during radiation therapy was predictive of radiation-induced lung toxicity in patients with non-small-cell lung cancer (Zhao et al. 2009). In animal experiments, long-term overexpression and activation of TGFß1 have been demonstrated in tissue as well (Johnston et al. 1995; Vujaskovic et al. 2002a). Thus, elevations in plasma TGFß1 months after radiation exposure appear to reflect the presence of significantly dysregulated wound healing in the irradiated tissues. In contrast, the absence of sustained elevations of circulating TGFß1 levels appear to reflect a more normal wound healing process. Thus, prolonged elevations of plasma TGFß1 following radiation exposure may be a useful means to identify patients at risk for late radiation-induced injury. Other investigators, however, have not found plasma TGFß1 to be a reliable identifier of patients at increased risk for normal tissue injury after cancer therapy (Barthelemy-Brichant et al. 2004; De Jaeger et al. 2004; Novakova-Jiresova et al. 2004). These discrepancies may be due to a number of factors, including differences in techniques used to measure TGFß1, differences in patient populations under study, differences in tumor type and burden, and the fact that these studies contain relatively small numbers of patients with treatment-related injury, thus the power to detect a difference between groups is not large (Anscher and Kong 2005).
5 The Role of Other Cytokines in Radiation-Induced Injury
A growing body of evidence points toward a complex web of protein interactions as being important in the pathogenesis of radiation injury (see Table 1 and Fig. 1). For example, Huang et al. (2002) have found that IL-7, a cytokine that enhances T cell function and IFN-γ production, inhibits both TGFß1 production and signaling, and protects against the development of bleomycin-induced pulmonary fibrosis. Fedorocko et al. (2002) showed that radiation exposure could increase cytokine production both directly (IL-6, TNF-α) and indirectly (GM-CSF), either by locally acting paracrine or endocrine effects or as a result of systemic effects of early proinflammatory mediators such as IL-1 or TNF-α. There is no doubt that protein production is a dynamic process, which will change as a result of cancer treatment. Hong et al. (2003) have documented temporal and spatial changes in the expression of proinflammatory cytokines (TNF-α, IL-1α, and IL-1ß) following thoracic irradiation in mice. Given the impact that radiation has on the expression of these and other proteins in tissue and that these changes in tissue protein expression might be reflected in changes in plasma protein levels, it is reasonable to postulate that it may be possible to quantify an individual patient’s inflammatory status by measuring candidate protein levels in the blood.
Table 1
Summary of the function of candidate proteins for profiling
Protein | Function |
|---|---|
IL-1ß | Inflammation, growth factor expression |
IL-5 | Proinflammatory |
IL-6 | Proinflammatory, decrease apoptosis of activated lung fibroblasts |
IL-7 | Proinflammatory |
IL-8 | Angiogenesis, leukocyte chemotaxis, and collagen synthesis |
IL-10 | Anti-inflammatory (decrease TNFα production, decrease upregulation of endothelial cell adhesion molecules) |
IL-13 | Proinflammatory |
MCP-1 | Inflammation, chemoattraction of monocytes |
MIP-1alpha | Antiproliferative |
PDGF BB | Angiogenesis, recruit smooth muscle cells |
VEGF | Angiogenesis and increased vascular permeability |
EGF | Epithelial cell motility, mitogenicity, and differentiation |
EGFR | Receptor for EGF, initial component of EGF signaling pathway |
NFkappaB | Pleotrophic gene transcription responses |
HIF-1 | Transcription factor for genes regulating angiogenesis |
TGF-alpha | Cell motility and proliferation |
FGF 2 | Angiogenesis and fibroblast proliferation |
MMP-1 | Degradation of collagen and extracellular matrix proteins |
MMP-2 | Matrix remodeling, growth factor release |
MMP-3 | Matrix remodeling, growth factor release |
MMP-13 | Matrix remodeling, growth factor release |
SMAD 2/3 | Signal transduction in the TGFß pathway |
IGF-1R | Binding of IGF-1 (re-epithelialization and granulation tissue formation) |
TNF-alpha | Growth factor expression, inflammation, matrix production, and remodeling |
TGFß1 | Profibrotic, immunosuppression, angiogenesis, and metastasis |
6 Using Other Markers to Predict Radiation-Induced Injury
In addition to TGFß1, several other proteins have been studied in humans to evaluate their potential as biomarkers for radiation-induced injury. Most of this work has been carried out in the lung. Of these, the most promising include interleukins (IL) 1α, IL-6, IL-8, IL-10, Krebs von den Lungen protein (KL-6, which is expressed mainly on type II pneumocytes and bronchiolar epithelial cells), soluble intracellular adhesion molecule (sICAM)-1, and surfactant proteins A and D (Kohno et al. 1992; Ishii and Kitamura 1999; Chen et al. 2001; Goto et al. 2001; Sasaki et al. 2001; Takahashi et al. 2001; Gridley et al. 2004; Hara et al. 2004, 2005; Chen et al. 2005; Matsuno et al. 2006). Of these, KL-6 is the most extensively studied, and has most consistently been correlated with the risk of radiation-induced lung injury (Fleckenstein et al. 2007a). As with TGFß1, more prospective studies with larger patient numbers will be required to confirm its value as a predictive marker for lung injury.
7 Chronic Inflammation as a Mediator of Radiation Injury
Epidemiologic evidence has also suggested a correlation between chronic inflammation and the development of malignancy at the inflamed site. Recent evidence points to a prolonged and progressive period of oxidative stress following the initial ionizing event in the development of radiation-induced lung injury. The underlying mechanism involves recruitment of inflammatory cells, as well as the expression of multiple mediators of inflammation, including cytokines, chemokines, and enzymes. Proinflammatory cytokines, such as the interleukins and tumor necrosis factor α, cause an influx of inflammatory cells and fibroblasts into the microenvironment (Johnston et al. 2004; Rube et al. 2005). These cells, primarily macrophages (Rubin et al. 1992; Vujaskovic et al. 2000), become stimulated to produce reactive oxygen species and additional proinflammatory and profibrotic cytokines (Fleckenstein et al. 2007a) (Fig. 1).
It is well established that tissue hypoxia is a potent stimulator of macrophage proliferation and activation. Fleckenstein et al. (Fleckenstein et al. 2007b) found a bi-phasic decrease in pulmonary perfusion following hemithoracic irradiation in rats, which correlated to the development of hypoxia, macrophage infiltration, and increased oxidative stress. Furthermore, in vitro studies lend support to the authors hypothesis that hypoxia stimulates TGFß1 and VEGF production by macrophages in an SOD-inhibitable manner.
Reactive oxygen species (ROS) functionally regulate transcription factors that also influence expression and activation of cytokines and growth factors (Sun and Oberley 1996). Furthermore, ROS play an important role in intracellular signaling, including activation of HIF-1a, NFkB, TGFß1, and a variety of other molecules found to play a role in radiation-induced injury (Schmidt-Ullrich et al. 2000). Over the past several decades, it has been shown that the free radical scavenging ability of endogenous antioxidants, most notably superoxide dismutase (SOD), acts as a cell-based protective mechanism. As the role of chronic oxidative stress in the development of radiation-induced lung injury has emerged, several investigators have attempted to restore the critical balance between chronic oxidative stress and antioxidant capacity through exogenous delivery of SOD. Several preclinical studies have shown mimics of SOD or liposomal-based delivery of SOD can mitigate injury when given after radiation and may even treat radiation-induced normal tissue injury once clinical symptoms become apparent (Epperly et al. 1999, 2000, 2008; Rabbani et al. 2007; Gauter-Fleckenstein et al. 2008, 2010; Borrelli et al. 2009). Thus, SOD-based therapy may hold potential therapeutic value in the treatment of radiation-induced lung injury.
Recent evidence suggests the importance of ROS/RNS generated by macrophages and tumor cells in the processes of initiation and progression of malignancy (Wink et al. 1998; Fukumura et al. 2006; Ridnour et al. 2006; Hirst and Robson 2007). Thus, it is likely that many, if not all, of the proteins involved in the development of radiation-induced normal tissue inflammation and fibrosis might also be involved in the generation of radiation-induced malignancy. In support of this idea, recently it has been shown that the SOD mimetic, MnTE-2-PyP (5+), a potent catalytic scavenger of reactive oxygen species that exhibited the ability to reduce radiation-induced lung injury (Gauter-Fleckenstein et al. 2008, 2010), also reduced tumor growth in a mouse model (Rabbani et al. 2009).
8 Candidate Proteins for Predicting Radiation Injury
While many proteins have been implicated in the pathogenesis of radiation-induced injury, few have been evaluated as possible predictors of predisposition to such injury. At the present time, not every protein implicated in inflammation, wound healing, fibrogenesis, or radiation response can be detected in the blood, owing to the lack of availability of reliable antibodies to these proteins. Thus, it is not yet possible to screen for alterations in expression of every potential candidate protein. In addition, multiple proteins and signaling pathways are involved in these processes, and reliable antibodies are not available to target every individual protein involved in each pathway. Nevertheless, the list of proteins below represent components of the major mechanisms and pathways currently thought to be involved in the response of cells to radiation (Schmidt-Ullrich 2003; Tsoutsou and Koukourakis 2006). This approach is likely to detect a profile of protein expression associated with an increased risk of radiation injury, if in fact one exists. The role of each of these candidate proteins, relevant to radiation injury, is summarized in Table 1.
9 Strategies and Potential Targets for Intervention
There are three primary approaches to intervention in the injury process, depending upon the timing of intervention relative to radiation exposure, and whether or not injury has developed (Moulder and Cohen 2007). These approaches are: protection or prophylaxis, mitigation, and treatment. Protection refers to treatments given before and/or during radiation. This is the most common strategy utilized in the clinic today and is illustrated by the use of the free radical scavenger amifostine in the prevention of injury following radiation to the head and neck (Brizel et al. 2000). Mitigation refers to therapies started after radiation exposure, but before overt injury is expressed, as exemplified by the use of angiotensin converting enzyme inhibitors to prevent renal injury (Moulder et al. 2003). Treatment refers to interventions begun after overt injury develops, an example of which would be the use of vitamin E and pentoxifylline to treat established radiation soft tissue fibrosis (Delanian et al. 2003) or the use of SOD mimics to treat symptomatic radiation-induced lung injury in rats (Gauter-Fleckenstein et al. 2010).
As we learn more about the specific molecular pathways involved in the process of radiation injury (Figs. 1, 2, 3), more targeted therapies are being studied as approaches to the prevention of radiation injury. Given the importance of the TGFß1 pathway in the pathogenesis of radiation injury, several investigators have demonstrated the efficacy of blocking TGFß1 in preventing radiation injury in animals (Flanders et al. 2002; Rabbani et al. 2003; Anscher et al. 2006, 2008). These agents, to date, have not been utilized in humans for this purpose. TGFß1 has also been demonstrated to work through Smad-independent pathways (Bierie and Moses 2006) and targeting one or more of these pathways may also prove to be an effective approach to prevention of radiation-induced injury. For example, one of these alternative pathways involves signaling via PI3-kinase and cAbl (Kharbanda et al. 1996a). The use of imatinib, which targets cAbl, has been shown to reduce the severity of bleomycin-induced lung injury (Daniels et al. 2004). In addition to TGFß1, other pathways have been demonstrated to be viable targets to inhibit the development of radiation-induced injury (Delanian et al. 1994; Hallahan and Virudachalam 1997; Adawi et al. 1998; Epperly et al. 1998; Arango et al. 2001; Kang et al. 2002; Vujaskovic et al. 2002b). Examples include reducing the level of TNFα expression using an antisense oligonucleotide strategy which reduced radiation-induced injury in mice (Zhang et al. 2008). In another approach, administration of the tyrosine kinase receptor inhibitor, Gefitinib, significantly reduced fibrosis scores and collagen levels at 5 months post-irradiation (Wang et al. 2008). The use of agents to scavenge free radicals to reduce oxidative/nitrosative stress has also been employed. In addition to the delivery of SOD itself or its mimetics, additional antioxidants have been examined. Taurine, which has been shown to exhibit antioxidant properties and inhibit TGFß1 mRNA expression, was shown to reduce lung tissue damage and hydroxyproline levels in mice (Robb et al. 2009). Genistein, an isoflavone found in soy with antioxidant and anti-inflammatory properties, reduced micronuclei formation and macrophage accumulation in mouse lungs after radiation (Para et al. 2009
 Mechanisms of Radiation Induced Injury
Mechanisms of Radiation Induced Injury
 Bioepidemiology of Multiple Primary Cancers
Consequences of Late Effects
Bioepidemiology of Multiple Primary Cancers
Consequences of Late Effects
 of the Pathophysiology Paradigm
of the Pathophysiology Paradigm
 of Normal Tissue TNM Toxicity Taxonomy: Scoring the Adverse Effects of Cancer Treatment
of Normal Tissue TNM Toxicity Taxonomy: Scoring the Adverse Effects of Cancer Treatment
 Analyses of RT-Induced Late Normal Tissue Injury Using Functional Imaging
Analyses of RT-Induced Late Normal Tissue Injury Using Functional Imaging




Related posts:
Mechanisms of Radiation Induced Injury
Bioepidemiology of Multiple Primary Cancers
Consequences of Late Effects
of the Pathophysiology Paradigm
of Normal Tissue TNM Toxicity Taxonomy: Scoring the Adverse Effects of Cancer Treatment
Analyses of RT-Induced Late Normal Tissue Injury Using Functional Imaging
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
