KEY POINTS
Anomalies of ventral induction of the fetal brain include a group of conditions that share in common an anomalous process of cleavage of the brain and formation of midline structures, encompassing a wide spectrum of severity. They are frequently associated with other malformations and genetic conditions.
Holoprosencephaly is one of the most severe disorders of ventral induction; it features incomplete separation of the cerebral hemispheres and has a very severe prognosis in most cases.
Agenesis of the corpus callosum is either complete or limited to the posterior portion; the anatomy is variable, but enlargement of the lateral ventricles and the absence of the cavity of the septum pellucidum are frequently present. The prognosis is largely influenced by the the association with other anomalies; isolated agenesis of the corpus callosum may be associated with a normal intellectual development, although the experience is limited.
Agenesis of the septum pellucidum is frequently a part of other, often severe malformations, including holoprosenephaly, gross hydrocephaly, and schizencephaly. Isolated agenesis of the septum pellucidum may be a normal variant, although it may be the only antenatal finding of septo-optic dysplasia, a condtiion that is usually associated with visual impairment and endocrine disfunction.
The ontogenesis of the cerebral midline is frequently referred to as the process of ventral induction. It takes place between the fifth week after conception and midgestation.1 Anomalies occurring during this period lead to the development of a wide range of malformations with a severity that is closely related with the time of occurrence. Beause ventral induction is closely related to facial development, many fetuses and children with prosencephalic disorders suffer from facial anomalies. A categorization of these anomalies is presented in Table 6–1. Disorders of prosencephalic formation, aprosencephaly and atelencephaly, will not be discussed in the following sections, as they are the extreme end of the spectrum of severity, and their occurrence is extremely rare. Aprosencephaly is the complete lack of development of the telencephalon and diencephalon, and in atelencephaly, the diencephalon is present but usually abnormal.2 In all prenatal reports, a different brain malformation was diagnosed in utero, and the definitive diagnosis was reached only after pathologic examination.3 Disorders of prosencephalic cleavage, holoprosencephaly and holotelencephaly, follow in the degree of severity. Disorders of prosencephalic midline development, agenesis of the corpus callosum, agenesis of septum pellucidum, and septo-optic dysplasia, usually have less severe presentations, but affected subjects may suffer from neurodevelopmental retardation and endocrinologic and visual disorders.
Disorders of prosencephalic formation Aprosencephaly Atelencephaly |
Disorders of prosencephalic cleavage Holoprosencephaly Holotelencephaly |
Disorders of prosencephalic midline development Agenesis of the corpus callosum, complete and partial Agenesis of the septum pellucidum Septo-optic dysplasia |
Holoprosencephaly is a genetically and phenotypically heterogeneous disorder involving the development of the forebrain and midface.
Arhinencephaly
Holoprosencephaly is seen in 1.0 to 1.7 per 10,000 births and terminations.4,5 The real incidence is probably higher than that, because the most severe forms of holoprosencephaly may result in early pregnancy loss,6 and minor forms will not be recognized at birth, thus escaping epidemiologic surveys.
Holoprosencephaly is commonly regarded as the result of a failure of cleavage of the prosencephalon. The prosencephalon is the most rostrad of the three primitive cerebral vesicles and gives rise to the cerebral hemispheres and diencephalic structures (including the neurohypophysis, thalami, and third ventricle). This differentiation process is thought to be induced by the precordal mesenchyma, which is probably also responsible for the differentiation of the median facial structures (forehead, nose, interorbital structures, premaxilla, and upper lip).
An interference with the activity of the prechordal mesenchyma would lead to defects in both the brain and midface. The cerebral anomalies are due to varying degrees of failure of cleavage of the derivatives of the prosencephalon, with incomplete division of the cerebral hemispheres and underlying structures. The facial anomalies encompass a broad range of defects that are due to aplasia or varying degrees of hypoplasia of the median structures (Table 6–2). Holoprosencephaly indeed fits the definition of a malformative sequence, featuring both cerebral and facial anomalies.
| Cyclopia | Single eye or single orbit Arrhinia with prosboscis |
| Ethmocephaly | Extreme hypotelorism Arrhinia with prosboscis |
| Cebocephaly | Orbital hypotelorism Prosboscis-like nose, no cleft |
| Median cleft | Orbital hypotelorism Flat nose |
| Agnathia-astomia | Hypoplasia or absence of the mandible Small or absent mouth Abnormal position of the ears |
Holoprosencephaly is a genetically heterogeneous condition involving at least 12 known loci on 11 chromosomes. The known holoprosencephaly-associated genes include Sonic hedgehog (SHH) on 7q36 (MIM 600725), ZIC2 on 13q32 (MIM 603073), SIX3 on 2p21 (MIM 603714), and TG-interacting factor (TGIF) on 18p11.3 (MIM 602630). Hereditary holoprosencephaly has been reported with autosomal dominant inheritance with variable penetrance (MIM 142945), autosomal recessive (MIM 236100), and X-linked recessive (MIM 306990).7,8 Holoprosencephaly is commonly associated with chromosomal abnormalities, mostly with trisomy 13 and triploidy.9,10,11 Syndromes that include holoprosencephaly among their manifestations are DiGeorge, Meckel, Kallmann, campomelic dysplasia, Hall-Pallister, and Vasadi, among others. There are no environmental teratogens known to cause this malformation in humans, but in animals the condition can be induced by veratrum alkaloids and radiation. Ingestion of salicylates in pregnancy has also been reported in relation to holoprosencephaly.
Holoprosencephaly is a continuum of malformations (Figure 6–1). A widely accepted classification recognizes three major varieties: the alobar, semilobar, and lobar type.9 Recently, a new variant, the middle interhemispheric variant, has been described,12,13,14 and the term holotencephaly has been suggested to describe cases with less pronounced derangement, with hemispheres incompletely cleaved but normal diencephalon.15 In the alobar variety, the most severe one, the interhemispheric fissure and the falx cerebri are totally absent, there is a single primitive ventricle, the thalami are fused on the midline, and there is absence of the third ventricle, neurohypophysis, and olfactory bulbs and tracts. The term arhinencephaly is indeed frequently used as a synonym of holoprosencephaly. In the semilobar variety, the two cerebral hemispheres are partially separated posteriorly, but there is still a single anterior ventricular cavity. In both the alobar and semilobar forms, the roof of the ventricular cavity, the tela choroidea, normally enfolded within the brain, may balloon out between the cerebral convexity and the skull to form a cyst of variable size, commonly referred to as the dorsal sac. With lobar holoprosencephaly, the anatomical derangement is much more subtle. In pathologic studies, this condition is usually described as a brain almost completely divided into two distinct hemispheres, with the only exception being a variable degree of fusion at the level of the cingulate gyrus and frontal horns of the lateral ventricles. The septum pellucidum is always absent. The olfactory bulbs and tracts and the corpus callosum may be absent, hypoplastic, or normal. An interesting aspect of lobar holoprosencephaly that has been described in studies employing magnetic resonance is the fusion of the fornices, which are seen as a solid fasicle running in the midline in the upper portion of the third ventricle.16

Pleomorphic facial anomalies are a part of the holoprosencephalic sequence17 (Table 6–2). Facial anomalies are usually encountered with the alobar and semilobar types, rarely with lobar holoprosencephaly or holotelencephaly. It should be stressed that infants with any kind of holoprosencephaly may have a normal face.9
Holoprosencephaly reflects a very early derangement of embryogenesis and is very frequently associated with other malformations.8 Apart from facial anomalies, which are considered a part of the malformative sequence, and other telencephalic anomalies, such as interhemispheric cysts and lipomas, holoprosencephaly has a striking association with two other conditions: chromosomal aberrations and Dandy-Walker malformation. The list of chromosomal aberrations found in patients with holoprosencephaly includes more than 35 conditions, but only 7 are frequently described: trisomy 13, del(13q), del(18p), trisomy 18, triploidy, dup(3p), and de1(7)(pter+q32).8,10,11 It has been estimated that over 60% of infants with trisomy 13 have holoprosencephaly; conversely, holoprosencephaly is associated with trisomy 13 in ~20% of cases. All varieties of holoprosencephaly are often associated with microcephaly, less frequently with macrocephaly, which is invariably due to internal obstructive hydrocephaly.
Holoprosencephaly with normal chromosomes has an estimated recurrence risk in siblings of 6%, but this calculation is derived from both truly sporadic events and hereditary conditions with 25% to 50% risk.8
The most valuable clue to the diagnosis is the demonstration of the single primitive ventricle (Figure 6–2).9,18,19 When present, the dorsal sac can be recognized (Figure 6–3). Depending on the degree of enfolding of the cortex over the ventricle, alobar holoprosencephaly may be further subdivided into a pancake, cup, and ball variety (Figure 6–4). Demonstration of facial anomalies such as cyclopia, hypotelorism, and median cleft lip strengthens the diagnosis of holoprosencephaly based on central nervous system (CNS) findings. Conversely, should any of the mentioned facial features be serendipitously encountered, a careful examination of the intracranial contents is recommended.
Figure 6–2.
Most cases of alobar holoprosencephaly seen around midgestation will appear like this: In an axial scan at the level of the thalami, there is no midline echo, and a crescent-shaped ventricular cavity is seen between the anterior frontal cortex and the posterior bulb-shaped thalami. The posterior fossa may be normal. (Reproduced, with permission, from Atlas of Obstetric Ultrasound, 2009. The Global Library of Women’s Medicine. www.glowm.com.)

Figure 6–3.
Multiplanar sonography of alobar holoprosencephaly in the midtrimester. (A) Median plane demonstrating the single ventricular cavity, which has a rim of cortex anteriorly and amply communicates posteriorly with a dorsal sac. (B) Axial scan at the level of the thalamus, demonstrating the crescent-shaped single ventricle and the absence of the midline in the anterior cortex. (C) In a slightly craniad axial plane than the previous one, the communication between the ventricular cavity and the dorsal sac is demonstrated. (Reproduced, with permission, from the Visual Encyclopedia of Ultrasound in Obstetrics and Gynecology, International Society of Ultrasound in Obstetrics and Gynecology, 2010, www.isuog.org.)

Figure 6–4.
In alobal holoprosencephaly, a dorsal sac is frequently seen on top of the ventricular cavity; the degree of development of the cortex is variable. (A) In some cases, it forms a thin rim at the base of the ventricles (pancake). (B) In other cases, it is partially enfolded on top of the ventricular cavity. (C) In still other cases, the ventricle is completely covered, and there is no dorsal sac; frequently, these cases are pathologically diagnosed as belonging to the semilobar variety. (Reproduced, with permission, from the Visual Encyclopedia of Ultrasound in Obstetrics and Gynecology, International Society of Ultrasound in Obstetrics and Gynecology, 2010, www.isuog.org.)

By using high-frequency transvaginal transducers, diverticulation of the forebrain can be demonstrated as early as the seventh week of amenorrhea. Indeed, by using this approach, a diagnosis of the alobar variety can be easily made at the onset of the second trimester,20 and in some instances even during the first trimester18,21 (Figure 6–5).
Figure 6–5.
Alobar holoprosencephaly in a fetus at 13 postmenstrual weeks. The most striking findings, the absence of the midline echo and the presence of a single rudimentary ventricular cavity, are well demonstrated in a vaginal scan. (Reproduced, with permission, from Tutschek B, PiluG. Virtual reality ultrasound imaging of the normal and abnormal fetal central nervous system. Ultrasound Obstet Gynecol. 2009;34(3):259–267.)

Timor-Tritsch et al22 used three-dimensional (3D) inversion rendering in the first as well as the early second trimester as an aid to diagnose holoprosencephaly. It was possible to discern between alobar and semilobar holoprosencephaly (Figure 6–6).
Figure 6–6.
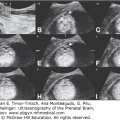
Pictorial table showing three-dimensional inversion rendering of the fluid in the ventricles of fetuses with holoprosencephaly in lateral, frontal, and superior views. (A) 9 weeks 2 days; (B) 10 weeks 2 days; (C) 13 weeks 5 days; (D) 14 weeks 5 days. (Reproduced, with permission, from Timor-Tritsch IE, Monteagudo A, Santos R. Three dimensional inversion rendering in the first and early second trimester fetal brain: Its use in holoprosencephaly. Ultrasound Obstet Gynecol. 2008;32:744–750.)




The ultrasonic findings of semilobar holoprosencephaly are very similar to the ones described for the alobar type. The diagnosis of the semilobar variety is suggested by the presence of well-developed occipital horns. Although lobar holoprosencephaly is amenable to antenatal identification, a specific diagnosis is difficult. The typical case will present with absence of the septum pellucidum and slightly enlarged and dysmorphic lateral ventricles (Figure 6–7). The major problem resides in the differential diagnosis between lobar holoprosencephaly, agenesis of the septum pellucidum, and other hydrocephalic conditions associated with secondary disruption of the septum pellucidum. Failure to demonstrate a well-developed corpus callosum, poorly formed frontal horns, and incomplete separation of the inferior frontal lobes favor the diagnosis of lobar holoprosencephaly.9,16,23,24 Recently, it has been suggested that color Doppler may be useful for a specific diagnosis. In lobar holoprosencephaly, the frontal horns are fused, and the anterior cerebral arteries run along the surface of the brain instead of coursing within the interhemispheric fissure. This sign has been defined as the serpent crawling under the skull.23 The middle interhemispheric variant of holoprosencephaly is intermediate between the lobar and alobar variety. When compared with the alobar variety, the frontal lobes are more differentiated than usual with recognizable frontal horns. Posteriorly, however, there is one single large and undifferentiated ventricular cavity with a hypoplastic diencephalon (Figure 6–8).
Figure 6–7.
Lobar holoprosencephaly. (A) The cavum septi pellucidi is absent, and the lateral ventricles appear significantly dysmorphic and fused from the level of the frontal horns to the bodies. (B) In the sagittal plane, the corpus callosum is not clearly visible, and only an irregular ridge of tissue is seen bridging between the hemispheres posteriorly (arrow). (C–E) Coronal sections are most useful for the diagnosis in that they demonstrate inferior fusion of the frontal lobes, poorly developed fused frontal horns, and communication between the bodies of the lateral ventricles. (Reproduced, with permission, from the Visual Encyclopedia of Ultrasound in Obstetrics and Gynecology, International Society of Ultrasound in Obstetrics and Gynecology, 2010, www.isuog.org.)

Figure 6–8.
Middle interhemispheric variant of holoprosencephaly in the axial (A), anterior coronal (B), and midcoronal plane (C). The frontal horns are well developed, and there is a partial formation of the interhemispheric fissure. However, the midcoronal plane reveals a common ventricular cavity with hypoplastic undivided thalami.

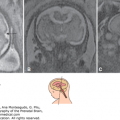
A specific diagnosis of holotelencephaly is difficult. We have always found this condition with severe obstructive ventricular dilation, as well as well-formed but fused frontal horns because of the absence of the septum pellucidum. In all our cases, as well as in some cases of the lobar type, the fornices are fused and are seen in the midline as a thick fasicle running from the anterior to the posterior commissure.16 In a midcoronal scan, the abnormal fornices result in a peculiar image: a small, round structure is seen in the midportion of the third ventricle (Figure 6–9). Fetal magnetic resonance imaging (MRI) may have a role in the elucidation of particularly difficult cases of lobar types or some of the nonclassical forms, such as the middle interhemispheric variant.13,25
Figure 6–9.
The appearance of a well-developed frontal cortex and ventricular system associated with obstructive dilation favors the diagnosis of holotelencephaly. The fornices (arrow) are fused and form a thick fascicle running in the floor of the ventricular cavity. (Reproduced, with permission, from Pilu G, Ambrosetto P, Sandri F, et al. Intraventricular fused fornices: A specific sign of fetal lobar holoprosencephaly. Ultrasound Obstet Gynecol. 1994;4[1]:65–67.)

Alobar holoprosencephaly with a large ventricular cavity and/or dorsal sac has an appearance similar to that of hydranencephaly. Although in our experience demonstration of the remains of the frontal cortex and/or facial anomalies has always allowed a clear distinction between these two entities, it goes without saying that, from a practical perspective, a diagnostic error would be anyhow uneventful, given the very poor prognosis of both conditions. Lobar holoprosencephaly must be distinguished from simple hydrocephaly with secondary disruption of the septum pellucidum.12 A midcoronal scan of the fetal head is the most important view to differentiate among these two conditions, because it allows identification of findings that are typical of lobar holoprosencephaly: the flat roof of the frontal horns and the possible presence of the fused fornices. Distinction between lobar holoprosencephaly and the absence of the septum pellucidum, isolated or associated with septo-optic dysplasia, is a much greater challenge, and the experience is limited. We have always found lobar holoprosencephaly associated with significant derangements of the anatomy of the hemispheres. The presence of well-formed and separated frontal horns and corpus callosum favors the diagnosis of absence of the septum pellucidum.12
In many cases, expert vaginal sonography can diagnose alobar and possibly semilobar forms during the first trimester.18,21 The lobar variety is probably difficult to identify prior to 16 to 18 weeks.
Alobar and semilobar holoprosencephalies are associated with profound distortion of intracranial architecture that should always be detected with a standard sonographic examination performed in the midtrimester. Findings associated with the lobar variety may be subtle. The cavum septi pellucidi, however, is always absent, with central fusion of the frontal horns. Identification of the absence of the septum pellucidum is not always easy.26
Alobar holoprosencephaly is lethal, although cases with long survival rates have been described. Semilobar holoprosencephaly is not necesserily lethal, but it is associated with extremely severe neurologic compromise. When these conditions are identified in utero, termination of pregnancy could be offered prior to viability. A conservative management is recommended in continuing pregnancies. The prognosis of lobar holoprosencephaly is uncertain. The available clinical data are limited. It has been reported that affected individuals may have a normal life span, but mental retardation and neurologic sequelae are common. Dysplasia of the aqueduct of Sylvius is presumably present in many cases, leading to obstructive hydrocephaly. The only available antenatal series includes five infants who were followed up after birth, and all had very abnormal developmental quotients. This series may be biased by the inclusion of fetuses with severe hydrocephaly and other associated anomalies.24
Fetal karyotype is mandatory when holoprosencephaly is discovered by ultrasound (US). Termination of pregnancy can be offered to parents with previable fetuses. Because alobar and semilobar holoprosencephalies are associated with a dismal prognosis, in the presence of severe hydrocephaly, US-guided cephalocentesis could be considered to avoid the risk of dystocia.