Fig. 1 a, b
A subtle cortical thickening of the distal lateral femoral cortex is evident on both radiographs, which display the knee joint and the hip joint, respectively. This highlights the importance of careful cortical scrutiny of the femoral cortex in patients radiographed for presumed articular disease
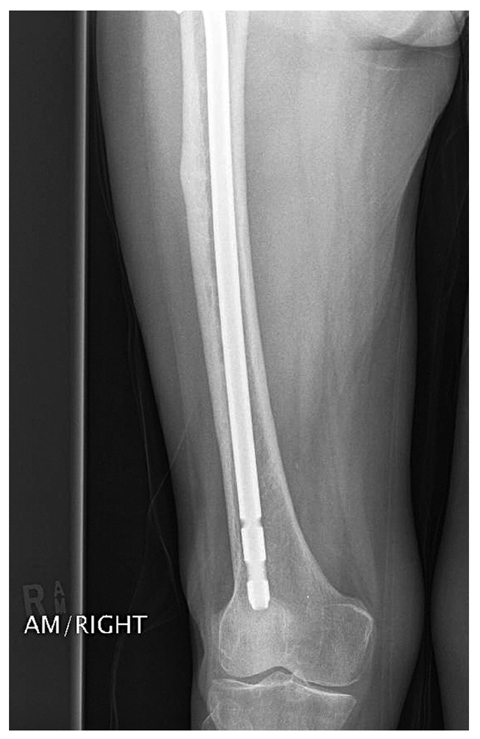
Fig. 2
Intramedullary nailing of the atypical femoral insufficiency from a shattered displaced fracture
Osteoporosis in the First Three Decades
In 2000, Glorieux and co-workers described a subtype of osteogenesis imperfecta (OI) with a propensity to such abundant hyperplastic callus after fracture that it bore a strong resemblance to osteosarcoma. They designated this particular complication as type V OI and considered the hyperplastic exuberant callus formation to represent a new form of brittle bone disease [8]. This form of the disease with hyperplastic callus can also lead to significant long-term morbidity [9].
Female Athlete Triad (Third Decade)
The female athlete triad comprises eating disorder such as anorexia nervosa, menstrual disorder (amenorrhea or oligomenorrhea) and osteopenia/osteoporosis [10]. „The female athlete triad” becomes a diagnostic consideration for the radiologist when stress fractures and serous atrophy of bone marrow are identified on MRI [11]. Serous atrophy of bone marrow on MRI demonstrates abnormal low signal in T1-weighted images, and high signal on T2-weighted or STIR images, and in advanced cases it can be associated with loss of subcutaneous fat and loss of fat in muscle septa. Stress fractures are usually an unequivocal finding on MRI, but might be difficult to identify in the presence of serous marrow change because the typical edema pattern silhouetting a fracture in normal bone marrow is masked by a serous marrow [12].
Osteomalacia and Rickets
Classically, the deficiency of vitamin D, essential for the absorption of calcium, has been the major cause of rickets in the child and osteomalacia in the adult, as a consequence of absence or delay in the mineralization of growth cartilage or newly formed bone collagen. Perhaps not as widely recognized is the development of rickets/osteomalacia as a consequence of a low serum phosphate and normal serum calcium. Two such conditions are X-linked hypophosphatemic rickets/osteomalacia and oncogenic osteomalacia. When present, the signs of rickets and osteomalacia in the low serum phosphate states are indistinguishable from the classic hypo-calcemic states, i.e., widened growth plate, metaphyseal cupping and fraying about joints in the child and Looser’s zones (a lucency that runs perpendicular to the cortex of bone, and is indistinguishable from the frequently encountered stress fracture) in the adult [13, 14]. However, there are some distinctive imaging signs that have been recently emphasized and described in these two conditions, along with clinical, genetic and biochemical advances.
X-linked Hypophosphatemic Osteomalacia
The condition is characterized by low tubular reabsorption of phosphate in the absence of secondary hyperparathyroidism. X-linked hypophosphatemia occurs in about 1 in 25,000 and is said to be the most common form of genetically induced rickets [15]. From an imaging standpoint, a curious and paradoxical finding in this condition is that in addition to Looser’s zones and femoral bowing there may be striking extraskeletal ossifications at sites of enthesis (mimicking a seronegative spondylitis or fluorosis) and occasionally intraspinal ossifications [16, 17].
Oncogenic Osteomalacia
Oncogenic osteomalacia, first described by Prader et al. in 1959 [18], is a paraneoplastic syndrome in which a bone or soft tissue tumor or tumor-like lesion induces hypophosphatemia and low vitamin D levels that reverse when the inciting lesion is resected.
Imaging Considerations
As elusive as the diagnosis of oncogenic osteomalacia appears to be for the clinician, it is more so for the radiologist unaware of the critical biochemical abnormalities of hypophosphatemia and hyperphosphaturia. The radiologist may consider the diagnosis of oncogenic osteomalacia if radiographs demonstrate Looser’s zones in adults or the classic signs of rickets in children in the absence of malnutrition or malabsorption. Once the presumptive clinical diagnosis of oncogenic osteomalacia is made, the search for the causative neoplasm begins. A recent review observed a range of 5 months to 14 years from the time of presentation to diagnosis [19]. Skeletal surveys and radioisotope bone scans have been the mainstays in the search for a clinically inapparent neoplasm. More recently, whole body MRI and indium III labeled octreotide scanning have been employed [20, 21]. Whatever the imaging modality used, any discovered lesion, however innocuous, should be considered causative of the syndrome and removed [22]. Tumors responsible for this syndrome may arise equally in bone or soft tissue with a predilection for the craniofacial region and extremities. A single case report and a single case from our institution has demonstrated cure being achieved by imaging-guided radiofrequency ablation of the suspected tumor, which pathologists now characterize as a phosphaturic mesenchymal tumor.
Renal Osteodystrophy
The connection between renal glomerular disease and bone disease was made a little over a hundred years ago [23], and the term renal osteodystrophy to describe the musculoskeletal complications of chronic renal failure was introduced in 1943 [24]. Renal osteodystrophy is the result of two major pathological processes that vary in severity: hyperparathyroidism from an excess of parathyroid hormone, and rickets or osteomalacia from a deficiency of 1,25-dihydrocholecalciferol, the renal hormone of vitamin D [25]. Dialysis and renal transplantation, the only life-sustaining and life-saving therapeutic options for chronic renal failure, modify the natural history of renal osteodystrophy, i.e., hyperparathyroidism and osteomalacia. Worldwide there are over one million patients on maintenance hemodialysis [26]. Since hyperparathyroidism is universal in chronic renal failure irrespective of imaging findings, it may be assumed that these patients have renal osteodystrophy. The development of a new disease state, amyloidosis, is a direct consequence of long-term dialysis in chronic renal failure. The problem is unresolved and on the rise with patients kept alive by dialysis for long periods of time while awaiting renal transplantation. Cameron refers to this as the „rise and rise of amyloidosis” [27]. Amyloid deposition is a consequence of B2 microglobulin, which is elevated to between 30 and 50 times the normal level in patients with chronic renal failure. The complication is almost universal in patients who have been on dialysis for 15 years or longer, tending to become evident after 8 years [27]. The most common location of these deposits is the carpal tunnel. Because primary hyperparathyroidism is diagnosed at a biochemical level, and more patients with renal failure are being sustained for longer periods of time on peritoneal or hemodialysis, brown tumors are now not uncommonly seen in poorly controlled patients on dialysis. Thus, in contradistinction to what was taught three decades ago, the practicing radiologist in the western world is more likely to encounter „brown” tumors more often as a manifestation of secondary rather than primary hyperparathyroidism.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


