Fig. 3.1
Regulation of bone mass with age in both sexes by growth hormone (GH), estrogen, androgen, FSH, parathyroid hormone (PTH), and loading. The increase in fracture risk is earlier in women than in men, and it is associated with both the higher level of FSH in peri- and post-menopause, and estrogen withdrawal
3.4.1 Growth Hormone
GH, secreted by the anterior pituitary, plays a major role in post-natal longitudinal bone growth owing to its ability to stimulate precursor cells in the epiphyseal cartilage [13]. This hormone also affects the mature skeleton by bone remodeling modulation [13]. These effects are mediated both directly through GH receptor and indirectly by the growth-promoting polypeptides, IGF-I and IGF-II [13]. IGF-I is synthesized mainly in the liver and the majority of it circulates to exert an endocrine action. It is also expressed by bone cells, acting as an autocrine–paracrine factor. Using different mouse models, it has been established that circulating IGF-I plays a pivotal role in the acquisition of bone mass [13]. GH acts on osteoblasts both directly or through IGFs, stimulating the proliferation as well as the synthesis of bone matrix proteins [14, 15]. GH also enhances osteoclastic activity by increasing local IGF-1 production by bone marrow stromal cells [16]. IGF-I, in turn, promotes bone resorption both directly and indirectly. In particular, osteoclasts possess IGF-I receptors [17], which mediate the direct effects of IGF-I. IGF-I also alters the RANKL/OPG ratio [18] in favor of osteoclastogenesis, decreasing OPG synthesis and increasing RANKL expression [18]. GH affects calcium metabolism by rising the levels of 1,25-OH-vitamin D, through the increase in PTH and IGF-I [13]. The importance of the GH/IGF axis arises from the demonstration that its perturbation has been implicated in growth defects as well as in the pathogenesis of osteoporosis [13]. In particular, several studies have reported reduced circulating levels of IGF in osteoporotic women [13]. Aging is associated with a decrease in the amplitude and frequency of GH production, ultimately leading to a reduction in hepatic IGF-1 synthesis [13].
3.4.2 Sex Steroid
The sex steroid hormones estrogen and androgen are powerful regulators of bone remodeling. They influence positively the growth, maturation, and preservation of the female and male skeletons. In particular, skeletal size and volume are similar in prepubertal girls and boys [19]. However, gender differences in bone growth become apparent during puberty, with men reaching higher peak bone mass. In fact, the male skeleton is characterized by larger size and stronger bones. These skeletal gender differences are attributed to a stimulatory androgen action on periosteal bone formation in men [19]. In women, estrogen favors a greater endocortical apposition. Testosterone and estrogen are crucial determinants in the maintenance of bone mass integrity during adulthood. Indeed, in both genders, gonadal failure is the most important factor for the development of bone loss [19].
Estrogen effects on bone cells. To understand how the differential effects of estrogen on bone might occur and how estrogen might interact with other age-related processes, it is important to review the three fundamental effects on bone metabolism.
a.
Estrogen inhibits the activation of bone remodeling and the initiation of new BMUs. In particular, withdrawal of estrogen is associated with increased apoptosis of osteocytes [20]. Recent studies show that serum estradiol levels are inversely associated with serum levels of sclerostin, the key inhibitor of Wnt signaling [21].
b.
Estrogen affects osteoclast formation and life span. In particular, it decreases the production of bone-resorbing cytokines, such as interleukin-(IL)-1, IL-6, TNF-α, M-CSF, and prostaglandins by osteoblasts [22].
Estrogen suppresses the activity of mature osteoclasts by direct, receptor-mediated mechanisms [20]. The hormone normally suppresses RANKL production by osteoblasts as well as T- and B lymphocytes [23, 24] and increases OPG production by osteoblasts [25], so that its deficiency leads to an alteration in the RANKL/OPG ratio that favors bone resorption.
Estrogen also increases the production of TGF-β by osteoblast precursor cells [26]. TGF-β induces apoptosis of osteoclasts [27]. Estrogen decreases osteoclastogenesis blocking RANKL/M-CSF-induced activator protein-1-dependent transcription by reducing c-Jun activity [28]. It also promotes osteoclast and their precursors’ apoptosis through the activation of Fas/Fas ligand signaling [7]. It favors expression of Fas ligand in osteoclasts and osteoblasts [22].
c.
Finally, estrogen is clearly important for the maintenance of bone formation. The effects of estrogen on osteoblasts and their progenitors may be stage specific. In particular, estrogen reduces the self-renewal of early mesenchymal progenitors [29] and the commitment of precursor cells to osteoblast at the expense of adipocyte lineage [30] and prevents osteoblast apoptosis [22]. Estrogen has also been shown to enhance osteoblast differentiation, [22] although data are variable and seem to depend on the system used. Very recently, oxytocin, a neurohypophyseal hormone, has been proposed as estrogen mediator of osteoblast differentiation [31].
Androgen effects on bone cells. Androgens have potent effects on osteoblast formation, and those are differential whether they act on trabecular or periosteal bone. While androgens maintain trabecular bone mass and integrity, they favor periosteal bone formation in men [19]. At the cellular level, testosterone increases the life span of osteoblasts by decreasing apoptosis [32]. Furthermore, androgens stimulate the proliferation of osteoblast progenitors and the differentiation of mature osteoblasts [32]. The molecular mechanisms of androgen action on bone cells are less well described than for estrogen. However, some evidence indicates that they favor osteoblast proliferation and differentiation by increasing TGF-β and responsiveness to FGF and IGF-II [32–34]. Androgens may also decrease osteoclast formation and bone resorption through increased production of OPG by osteoblasts [35]. The net result is that testosterone favors bone formation. Moreover, testosterone may influence different stages of osteoblast differentiation and may act on osteoblasts differently than estrogen at various skeletal localizations. This last aspect of testosterone on bone formation is also important for the skeletal sexual dimorphism [19].
3.4.3 Follicle-Stimulating Hormone
Follicle-stimulating hormone (FSH) has traditionally been identified with ovarian folliculogenesis and estrogen production. However, it is now known that the peri- and post-menopausal spike in FSH stimulates osteoclastogenesis and bone resorption [36]. During the late peri-menopause, even before estrogen has declined, FSH levels increase. Accompanying this rise, there occurs a quick elevation in the markers of bone resorption and an increase in trabecular damage that deteriorates bone and increases fracture risk [37]. Approximately half of the bone loss over a typical woman’s lifetime occurs within the first five years of menopause. Interestingly, much of this loss occurs before significant estrogen declines [37]. The demonstration that FSH effects occurred independently of estrogen came from the finding that haploinsufficiency of FSH in mice resulted in a high bone mass; this increase occurs even though these mice have normal levels of estrogen. FSH stimulates osteoclast formation from its precursors in the presence of RANKL through the Gi2α-coupled FSH receptor, present on both precursors and mature osteoclasts [36]. FSH also stimulates monocytes to produce IL-1β, which in turn activates osteoclasts for bone resorption [37]. Furthermore, the hormone stimulates TNF-α production from the bone marrow precursor macrophages and granulocytes [38]. Likewise, FSH may be responsible for a surge in TNF-α produced in T cells during estrogen deficiency that contributes to bone loss.
3.4.4 Parathyroid Hormone
PTH is the principal regulator of calcium homeostasis and accordingly of bone remodeling since the skeleton represents the major reservoir/deposit of calcium. In response to hypocalcemia, parathyroid glands secret PTH, which acts on different peripheral targets in order to restore normocalcemia. It favors calcium release from bone increasing osteoclast activity. In kidneys, PTH promotes calcium resorption as well as activation of 1,25-OH-vitamin D, which enhances gut absorption of the ion. PTH activates the PTH1 receptor, leading to a wave of transcriptional responses that produce/modulate secretion of molecules exerting both catabolic and anabolic effects on bone. The PTH catabolic effect can both directly and indirectly activate the bone resorptive processes via the stimulation of osteoclast activity [6, 39]. Indeed, it raises the RANKL/OPG ratio by increasing RANKL production and decreasing OPG expression in osteoblasts, thus promoting indirectly osteoclast formation and activity [6]. It has been reported that MCP-1 can also mediate PTH catabolic effect through the recruitment of osteoclast precursors [5]. PTH exerts anabolic effects by direct and indirect mechanisms, resulting in higher bone mass, higher proliferation of bone marrow cells, and deeper extent of osteoblastic differentiation. Indeed, PTH directly enhances IGF-I synthesis by osteoblast [40] and attenuates the adipogenic effect of peroxisome proliferator-activated receptor-γ (PPARγ) favouring osteoblast differentiation [41]. PTH indirectly suppresses the expression of sclerostin, an inhibitor of the Wnt-β-catenin pathway responsible for osteoblastogenesis, thus leading to the increased osteoblast number and activity [42]. T cells expressing PTH1 receptor may be involved in both anabolic and catabolic effects of PTH through CD40L, a surface molecule of activated T cells that induced CD40 signaling in stromal cells [43]. In particular, the deletion of CD40L on T cells blunts the catabolic activity of PTH in bone by decreasing bone marrow stromal cell number, RANKL/OPG production, and osteoclastogenesis, whereas the T cell–mediated anabolic effect of PTH involves Wnt signaling activation in osteoblasts. In the last years, drugs mimicking PTH anabolic effects have been developed and are used as therapeutic strategy in osteoporosis management.
3.4.5 Wnt/β-Catenin Signaling
Another pathway named Wnt signaling has recently emerged to be important for bone development, homeostasis, and regeneration. Studies with animal models have been shown that changes in Wnt signaling contribute to age-related bone loss [44], and mechanical loading upregulates Wnt signaling in mesenchymal stem cells [45], suggesting that the combination of reduced Wnt signaling and decreased mechanical stimulation with age may contribute to the age-related decline in bone formation. Wnt signaling comprises several ligands, receptors, and inhibitors with bone-specific functions [46]. Activation of Wnt/β-catenin signaling occurs upon binding of Wnt to the 7-transmembrane domain-spanning frizzled receptor and low-density lipoprotein receptor–related protein 4, 5, and 6 (LRP4/5/6) co-receptors. This event prevents an intracellular protein complex consisting of Axin, GSK3ß, and APC from tagging ß-catenin for degradation [46]. As a result, ß-catenin accumulates in the cytosol and can translocate into the nucleus, where it interacts with Tcf/Lef transcription factors, leading to the expression of master genes for osteoblastogenesis. In particular, ß-catenin accumulation favors mesenchymal stem cell commitment for an osteogenic fate [47] and is needed to both promote early osteoblast proliferation and differentiation and suppress osteoclastogenesis [48].
There are several feedbacks which inhibit Wnt signaling, mediated by secreted molecules [46]. These soluble inhibitors include sclerostin, which antagonizes Wnt binding to LRP4-6 co-receptors, the Dickkopfs (Dkks), secreted frizzled-related proteins (sFrps), frizzled-related protein (Frzb), Wnt-1-induced secreted protein (WISE), Wnt inhibitory factor-1 and factor-2 (Wif-1, Wif-2) [46]. The secreted inhibitors, Dkk1 and sclerostin, are particularly interesting, as their increased expression has been demonstrated in many bone-associated diseases. In fact, they are targeted by therapeutic antibodies, which seem to be efficacious and safe in clinical settings.
Dkk1 suppresses Wnt signaling by forming a ternary complex with Lrp5/6 and Kremen (Krm)1/2 [49]. Transgenic mice expressing Dkk1 had fewer osteoblasts and severe osteopenia, whereas deletion of a single Dkk1 allele increased bone mass [49]. Dkk1 also switches mesenchymal stem cells to differentiate toward adipocytes. Moreover, Dkk1 affects the RANKL/OPG axis, in particular Dkk1 blockade leads to the overexpression of OPG [49]. On the contrary, Dkk1 treatment reduced the osteoblast expression of OPG and increased RANKL levels, thus promoting osteoclast formation [49].
The other key inhibitor, sclerostin, the product of SOST gene, is prominently produced by osteocytes embedded in newly formed bone [50]. Mutation or deletion of SOST leads to two rare bone sclerosing dysplasias, as sclerosteosis and van Buchem disease [50]. Sclerostin addition to osteogenic cultures inhibited proliferation and differentiation of mouse and human osteoblastic cells [50]. Moreover, sclerostin was shown to decrease the life span of osteoblasts by stimulating their apoptosis [50]. Mechanical loading decreased SOST mRNA and sclerostin levels, while unloading increased SOST mRNA expression in vivo. Interestingly, SOST knockout mice do not exhibit bone loss after unloading nor canonical Wnt signaling is altered [51]. Several systemic and local factors have been suggested as possible regulators of SOST/sclerostin expression by osteocytes. PTH has been shown to inhibit its expression both in vitro and in vivo [42], whereas BMP2, BMP4, and BMP26 stimulate SOST expression in osteoblastic cells [50].
3.4.6 Bone Morphogenetic Proteins
BMPs are also important regulators of skeletal development and maintenance and are produced by osteoprogenitor cells, osteoblasts, chondrocytes, and platelets [52]. Several clinical studies demonstrated the efficacy of BMPs in accelerating bone regeneration and fracture healing [52]. The osteogenic potency of the BMPs requires a local and controlled delivery. Moreover, for clinical use of BMPs, their short half-life time should be taken into account. Several delivery systems have been developed to overcome this limitation. BMPs bind to receptor complexes and activate Smad and MAPK pathways, which upregulate the expression of Runx2 and Osterix, thus promoting osteoblastogenesis and bone formation [53]. However, BMP2-4 ligands direct osteoblasts to reduce bone mass via BMPR1A, in part by upregulating sclerostin expression and supporting osteoclastogenesis through the RANKL-OPG pathway. However, this occurs during embryonic bone development [54]. The activity of BMPs is locally regulated by a number of antagonists, which are also important for bone remodeling and may be potential therapeutic targets for osteoporosis. BMP antagonists can be subdivided into molecules that act extracellularly or intracellularly. Extracellular secreted polypeptides, including noggin, chordin, gremlin, twisted gastrulation, and so on, can inhibit the signals by binding BMPs, thereby preventing them to bind their receptors [52], whereas intracellular antagonists intervene with the activation of R-Smads and/or facilitate their proteasomal degradation.
3.4.7 Mechanical Forces
Mechanical loading has prominent influences on bone remodeling. Disuse or lack of loading causes an acceleration of bone turnover, with osteoclast resorption dominating osteoblast formation with the result of a substantial bone mass loss. Disuse of bone (low stress) causes reduced bone formation on periosteal surfaces and increased bone resorption and turnover on endocortical and trabecular surfaces. This type of bone loss has also been observed in astronauts who spend extended periods of time in the microgravity environment of a space station or shuttle. Also, the overuse of bone can increase bone turnover; in fact, the excessive load stimulates bone turnover whose role is to replace and repair damaged bone tissue and this event provides the involvement of osteoclasts which target regions of microdamage and remove compromised bone. The damaged tissue will be then replaced by new bone. If damage accumulates and bone cannot effectively be repaired, microcracks may develop and propagate to form a stress fracture. Overuse injuries are often classified as stress or fatigue fractures and occur as a result of microdamage accumulation and loss of bone stiffness and strength. Mechanical loading influences bone remodeling following a U-shaped curve: As mentioned before, an increase in bone remodeling activity corresponds to a decrease in bone mass. Thus, the central part of the U represents the physiological range in which bone turnover is minimized. The unloading for disuse or microgravity as well as overloading are at the border of the U, indicating a bone remodeling increase. Periosteal bone formation, on the other hand, steadily enhances with increased loading, partially counteracting bone loss in overuse conditions (Fig. 3.2).
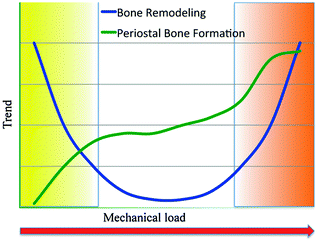
Fig. 3.2
Remodeling increases in states of disuse (yellow) and overuse (orange).A physiological window falls between the extreme loading conditions, in which bone turnover is low and bone mass is stable. Bone formation at the periosteal bone surfaces is also affected by mechanical loading, but the relationship is different. Periosteal bone formation is very low in states of disuse and increases with increasing mechanical stimulus partially offsetting the bone loss induced by the increased bone remodeling
The increased bone turnover both in disuse and in overuse events is correlated with osteocyte apoptosis [55




Related posts:



Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
