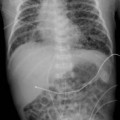
Challenge
Case 157
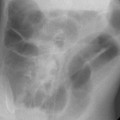
Answers: Case 157
Van der Knaap M.S., Valk J. Congenital muscular dystrophies. In Magnetic resonance of myelination and myelin disorders, ed 3, New York: Springer Berlin Heidelberg; 2005:451-468.
Case 158
Patel S., Barkovich A.J. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol. 2002;23:1074-1087.
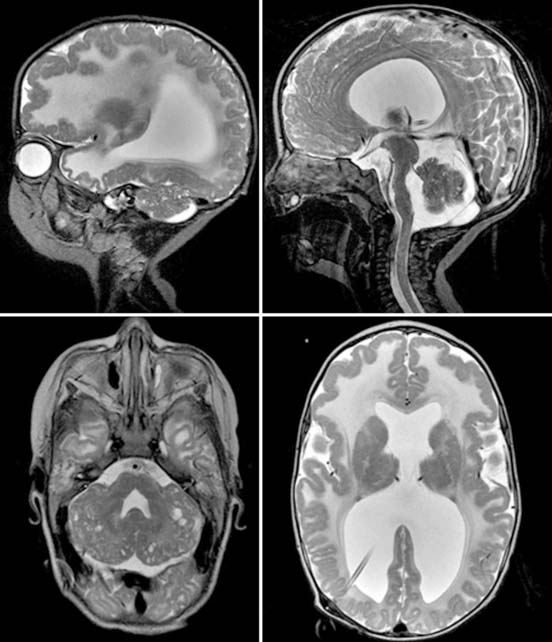
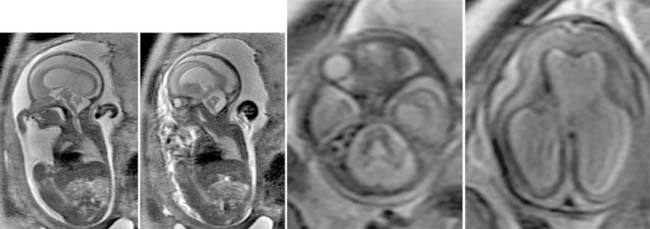
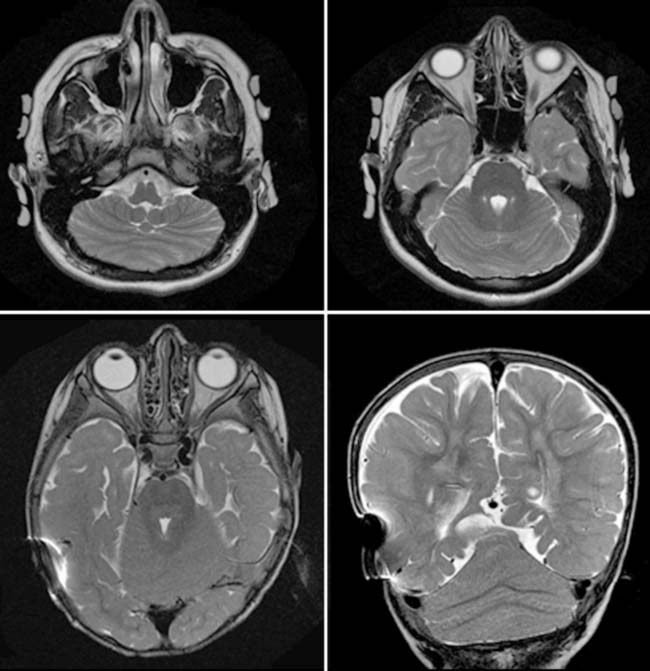
Case 159
Answers: Case 159
Poretti A., Boltshauser E., Loenneker T., et al. Diffusion tensor imaging in Joubert syndrome. AJNR Am J Neuroradiol. 2007;28:1929-1933.
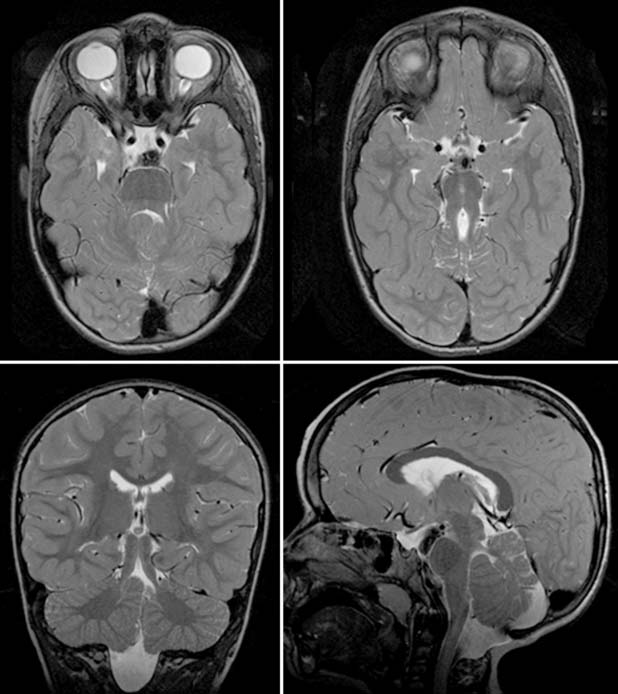
Case 160
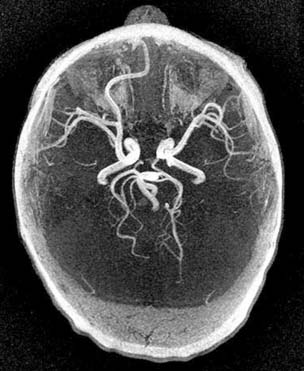
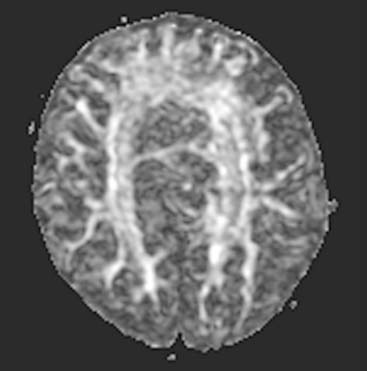
Answers: Case 160
Case 161
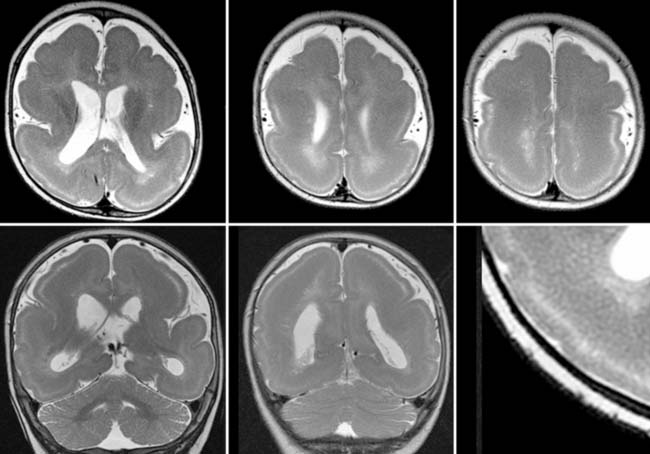
Answers: Case 161
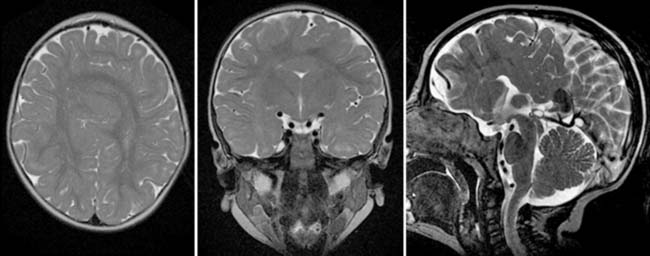
Case 162
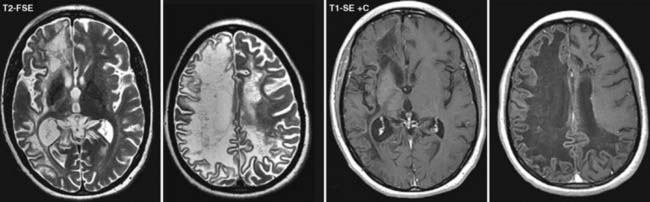
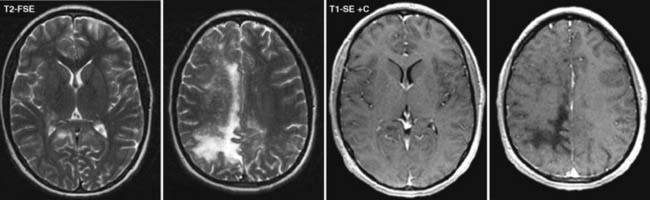
Answers: Case 162
Diagnosis: Progressive Multifocal Leukoencephalopathy
Case 163
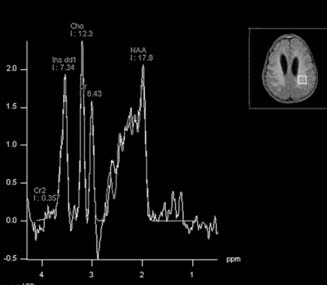
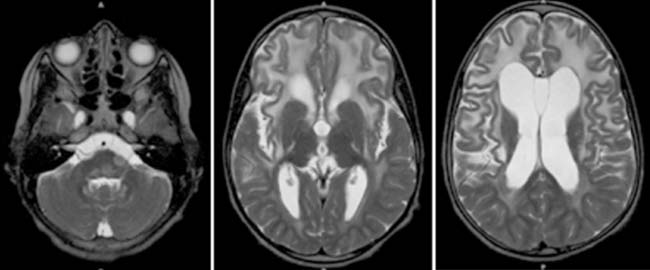
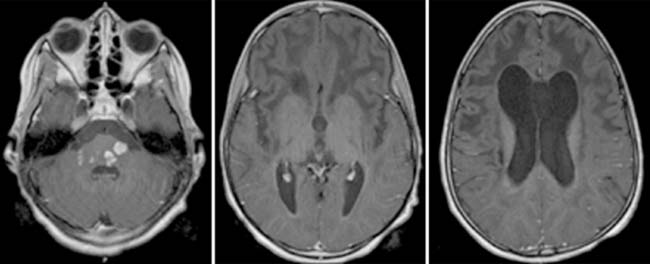
Answers: Case 163
Diagnosis: Alexander Disease
Van der Knaap M.S., Valk J. Congenital muscular dystrophies. In Magnetic resonance of myelination and myelin disorders, ed 3, New York: Springer Berlin Heidelberg; 2005:416-441.
Case 164
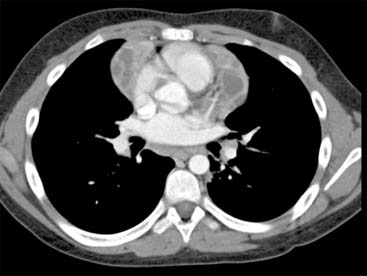
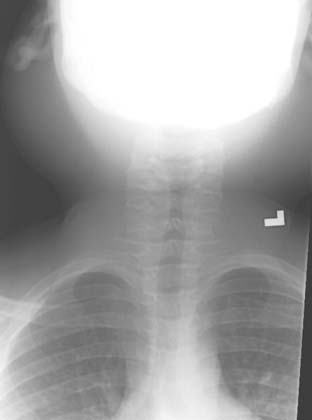
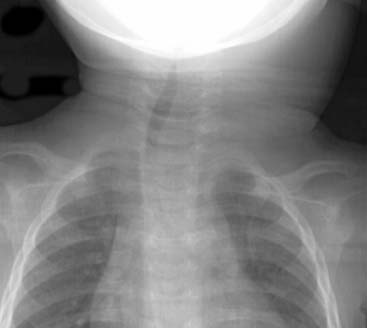
Answers: Case 164
Diagnosis: Positive Human Immunodeficiency Virus: Parotitis and Thymic Cysts
Kuhn J.P., Slovis T.L., Haller J.O. Caffey’s pediatric diagnostic imaging, ed 10, Philadelphia: Mosby; 2004:1184-1189.
Case 165
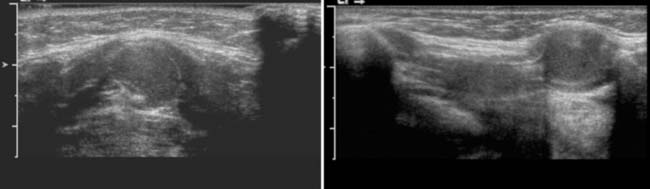
Answers: Case 165
Diagnosis: Thyroglossal Duct Cysts
Tunkel D.E., Domenech E.E. Radioisotope scanning of the thyroid gland prior to thyroglossal duct cyst excision. Arch Otolaryngol Head Neck Surg. 1998;124:597-599.
Case 166
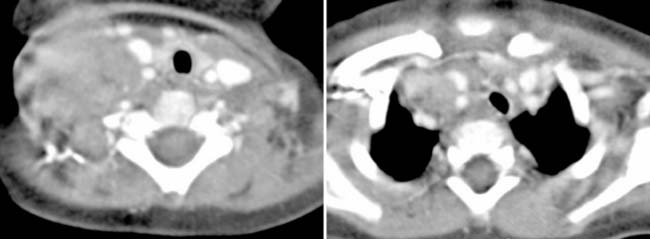
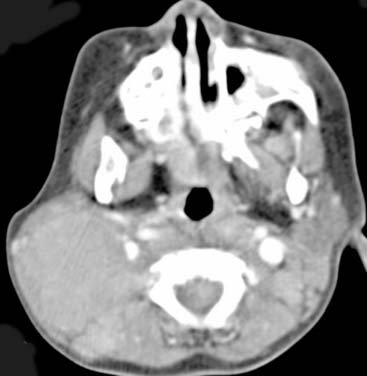
Answers: Case 166
Diagnosis: Rosai-Dorfman Disease
Rosai J., Dorfman R.F. Sinus histiocytosis with massive lymphadenopathy: a pseudolymphomatous benign disorder. Cancer. 1972;30:1174-1188.
La Barge D.V.3rd, Salzman K.L., Harnsberger H.R., et al. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): imaging manifestations in the head and neck. AJR Am J Roentgenol. 2008;191(6):W299-W306.
Case 167
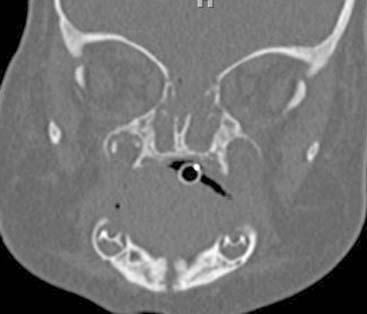
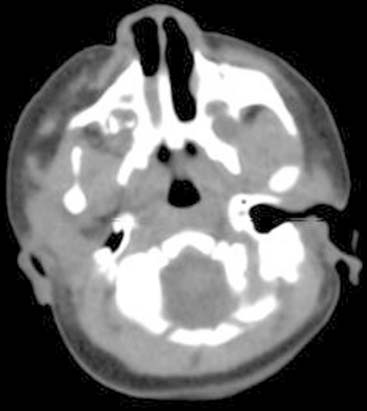
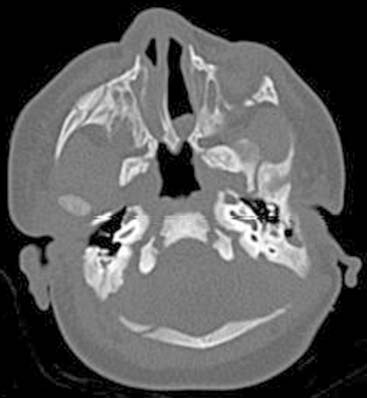
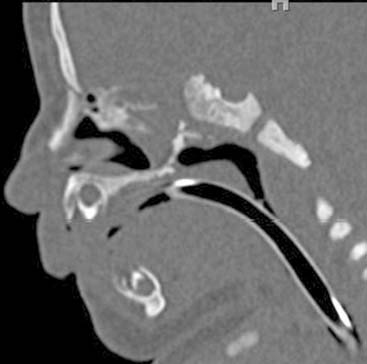
Answers: Case 167
Diagnosis: Choanal Atresia
Valencia M.P., Castillo M. Congenital and acquired lesions of the nasal septum: a practical guide for differential diagnosis. Radiographics. 2008;28:205-223.
Case 168
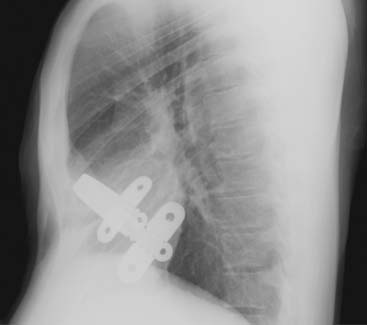
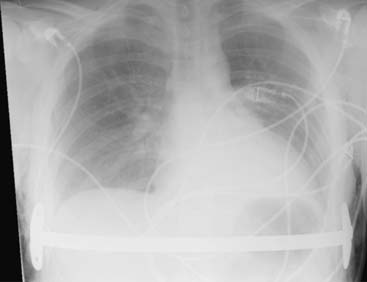
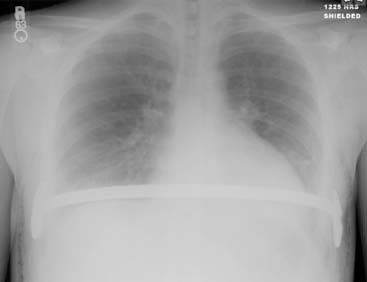
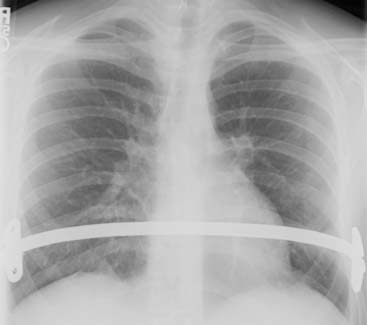
Answers: Case 168
Diagnosis: Pectus Excavatum Repair Complication
Vegunta R.K., Pacheco P.E., Wallace L.J., et al. Complications associated with the Nuss procedure: continued evolution of the learning curve. Am J Surg. 2008;195(3):313-316.
Case 169
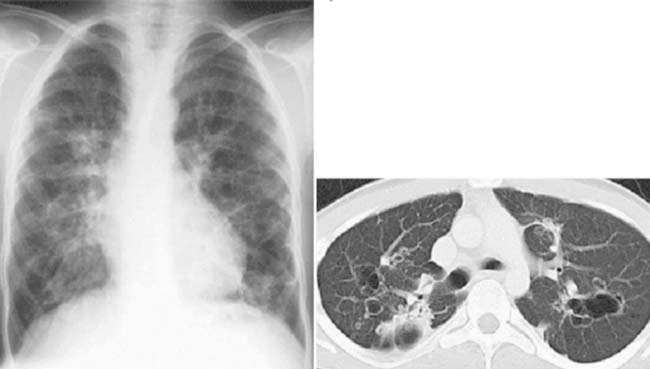
Answers: Case 169
Kuhn J. Larynx and cervical trachea. In: Kuhn J.P., Slovis T.L., Haller J.O., editors. Caffey’s pediatric diagnostic imaging. ed 10. Philadelphia: Mosby; 2004:812-813.
Effman E. Pulmonary neoplasms, tumor-like conditions and miscellaneous diseases. In: Kuhn J.P., Slovis T.L., Haller J.O., editors. Caffey’s pediatric diagnostic imaging. ed 10. Philadelphia: Mosby; 2004:1129-1130.
Case 170
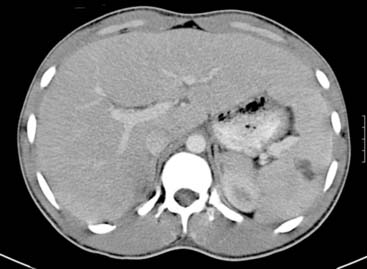
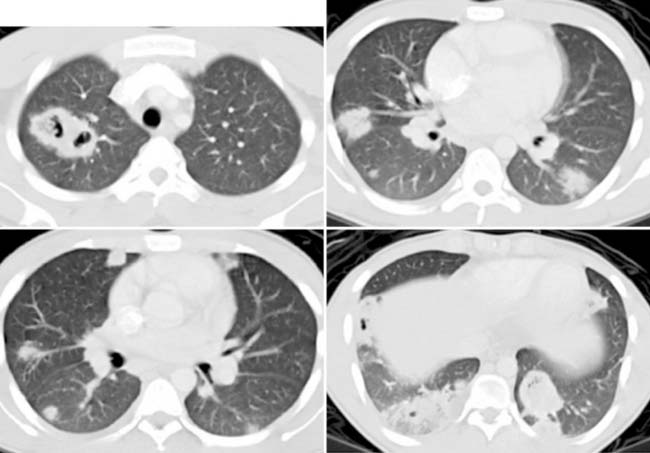
DeSena S., Rosenfeld D.L., Santos S., et al. Jugular thrombophlebitis complicating bacterial pharyngitis (Lemierre’s syndrome). Pediatr Radiol. 1996;26:141-144.
Lustig L.R., Cusick B.C., Cheung S.W., et al. Lemierre’s syndrome: two cases of postanginal sepsis. Otolaryngol Head Neck Surg. 1995;112:767-772.
Case 171