Introduction
Collagen vascular diseases (CVDs) are a heterogeneous group of immunologically mediated inflammatory disorders that may involve various organs. They often affect the lungs, mediastinum, and pleura, but the frequency of pulmonary involvement varies according to the specific disease. Additionally, medical treatment may lead to lung infection or drug reaction. Thus imaging is essential in the management of patients with CVD, with early diagnosis and intervention reducing patient morbidity and mortality.
General Aspects of Collagen Vascular Diseases
Collagen Vascular Diseases That Usually Involve the Chest
The defined CVDs that usually involve the lung are rheumatoid arthritis (RA), progressive systemic sclerosis (PSS), systemic lupus erythematosus (SLE), polymyositis (PM), dermatomyositis (DM), mixed connective tissue disease (MCTD), and Sjögren syndrome (SS).
Thoracic Manifestations With Greatest Clinical Impact in Patients With Collagen Vascular Disease
The two thoracic manifestations with the greatest clinical importance in patients with CVD are interstitial lung disease (ILD) and pulmonary arterial hypertension (PAH), which are the major contributors to mortality and morbidity in this patient group.
Best Imaging Techniques and Protocols for Assessing Patients With Collagen Vascular Disease
High-resolution CT (HRCT) is the reference standard technique to image pulmonary diseases in patients with CVD. Following the development and widespread availability of multidetector CT (MDCT) scanners capable of acquiring near-isotropic data throughout the entire thorax in a single breath-hold, two general approaches are available for performing HRCT. The first and more traditional method entails obtaining axial thin-section (0.625- to 1.5-mm slice thickness) HRCT images spaced at 10- to 20-mm intervals throughout the lungs. The second method uses the ability of MDCT scanners to provide volumetric single breath-hold datasets, allowing spaced, contiguous, and/or overlapping HRCT images to be reconstructed. With MDCT, volumetric data enables multiplanar thin-section HRCT reconstruction, which facilitates the evaluation of the distribution of diffuse lung diseases and the application of postprocessing techniques such as maximum intensity projection, minimum intensity projection, and software that uses volumetric data for quantification of features in the lungs and airways.
Interstitial Pulmonary Diseases in Patients With Collagen Vascular Disease
Most Common Interstitial Pattern
CVD may be associated with various patterns of ILD, and the relative frequency with which these patterns occur varies according to the type of CVD, demographics, and clinical characteristics of those affected ( Table 23.1 ).
| DISEASE | UIP | NSIP | OP | LIP | DAD | HEMORRHAGE | AIRWAY DISEASE |
|---|---|---|---|---|---|---|---|
| Rheumatoid arthritis | +++ | ++ | ++ | + | + | – | +++ |
| Progressive systemic sclerosis | + | +++ | + | – | + | – | – |
| Dermatomyositis/Polymyositis | + | +++ | +++ | – | ++ | – | – |
| Sjögren syndrome | + | ++ | – | ++ | + | – | + |
| Mixed connective tissue disease | + | ++ | + | – | – | – | – |
| Systemic lupus erythematosus | + | ++ | + | + | ++ | +++ | – |
a Symbols in columns indicate the frequency with which the feature or pattern occurs, with + indicating the lowest, +++ indicating the highest frequency, and – indicating absence of the feature or pattern.
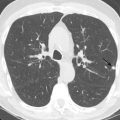
Nonspecific interstitial pneumonia (NSIP) is now recognized as a distinct clinicopathologic entity and has been shown to be the most common histologic pattern of ILD in patients with CVD. NSIP encompasses a broad spectrum of histologic features with varying degrees of inflammation and fibrosis, and the temporal uniformity of these pathologic features is the most distinguishing feature from usual interstitial pneumonia (UIP). NSIP is divided into cellular NSIP, which is characterized by inflammation without a substantial fibrotic component, and fibrotic NSIP, in which fibrosis is the dominant finding.
The most common features of NSIP on HRCT are ground-glass opacities, reticulation (usually representing fine fibrosis), and traction bronchiectasis or bronchiolectasis, with a symmetric basal predominant distribution. A lesser degree of involvement of the parenchyma immediately adjacent to the pleura (subpleural relative sparing) can also be seen ( Fig. 23.1 ) and is a distinguishing feature. Other findings include architectural distortion and lower lobe volume loss. Honeycombing is uncommon and occurs primarily in those with advanced fibrosis.

Accurate Detection of Pulmonary Interstitial Disease and Its Importance
Pulmonary alterations (physiologic or anatomic) may be the first manifestations of CVD. Accurate detection of pulmonary involvement in CVD has important therapeutic and prognostic implications because prompt treatment may lead to improved outcomes. Other radiologic findings suggestive of underlying CVD include esophageal dilation, pleural and pericardial effusions and thickening pulmonary arterial enlargement, and osteoarticular disease.
Although the prognosis of NSIP is usually better than that of UIP, NSIP is associated with substantial mortality; nearly 20% of patients die within 5 years after diagnosis. Patients with cellular NSIP have a more favorable prognosis than those with the fibrotic type. Individuals with CVD-associated ILD usually have a better prognosis than those with idiopathic ILD. Therefore evidence of an underlying CVD should be sought at the initial diagnosis of biopsy-proven NSIP and during the follow-up period, even in the absence of systemic symptoms.
Interstitial Pneumonia With Autoimmune Features and When This Diagnosis Should Be Considered
Some individuals with a diagnosis of idiopathic interstitial pneumonia (IIP) have clinical features that suggest an underlying autoimmune condition but do not meet recognized diagnostic criteria for any CVD. Many terms have already been proposed to describe this process, such as undifferentiated CVD-associated ILD (UCVD-ILD), lung-dominant CVD, and autoimmune-featured ILD.
To achieve consensus about the nomenclature and classification criteria for patients with IIP and features of autoimmunity, the European Respiratory Society/American Thoracic Society Task Force on Undifferentiated Forms of Connective Tissue Disease–Associated Interstitial Lung Disease was formed. The task force agreed on the term interstitial pneumonia with autoimmune features (IPAF) for these patients and proposed some classification criteria ( Box 23.1 ), which are centered around three central domains: (1) a clinical domain with specific extrathoracic features; (2) a serologic domain with specific circulating autoantibodies; and (3) a morphologic domain with specific HRCT, histopathologic, and multicompartment features. To be classified as having IPAF, a patient must meet all the a priori requisites and have at least one feature from at least two of the three domains.
- 1.
Presence of an interstitial pneumonia (by HRCT or surgical lung biopsy)
and
- 2.
Exclusion of alternative causes
and
- 3.
Absence of criteria for a defined CVD
and
- 4.
At least one feature from at least two of these domains:
- a.
Clinical domain: distal digital fissuring or tip ulceration, inflammatory arthritis or polyarticular morning joint stiffness ≥60 min, palmar telangiectasia, Raynaud phenomenon, unexplained digital edema, unexplained fixed rash on the digital extensor surfaces (Gottron’s sign)
- b.
Serologic domain: ANA ≥1 : 320, diffuse, speckled, homogeneous patterns or ANA nucleolar pattern or ANA centromere pattern; rheumatoid factor ≥2× upper limit of normal; anti-CCP, anti-dsDNA, anti-Ro (SS-A), anti-La (SS-B), antiribonucleoprotein, anti-Smith, antitopoisomerase (Scl-70), anti-tRNA synthetase, anti-PM-Scl, anti-MDA-5
- c.
Morphologic domain:
- 1)
HRCT patterns: NSIP, OP, NSIP with OP overlap, LIP
- 2)
Histopathology patterns: NSIP, OP, NSIP with OP overlap, LIP, interstitial lymphoid aggregates with germinal centers, diffuse lymphoplasmacytic infiltration
- 3)
Multicompartment involvement (in addition to IIP): unexplained pleural effusion or thickening, unexplained pericardial effusion or thickening, unexplained intrinsic airway disease, unexplained pulmonary vasculopathy
- 1)
- a.
ANA, Antinuclear antibody; CVD, collagen vascular disease; HRCT, high-resolution computed tomography; IIP, idiopathic interstitial pneumonia; LIP, lymphoid interstitial pneumonia; NSIP, nonspecific interstitial pneumonia; OP, organizing pneumonia.
Patients with IPAF do not have a classifiable CVD, but are distinct from other individuals diagnosed with IIP. Some of these patients will progress over time to a defined CVD. Prospective studies are still required to validate the proposed classification criteria and assess the natural history and clinical implications of a classification of IPAF.
Pulmonary Hypertension
Collagen Vascular Diseases Usually Associated With Pulmonary Arterial Hypertension
PAH is defined by a mean resting pulmonary artery pressure ≥25 mm Hg and a pulmonary capillary wedge pressure ≤15 mm Hg. Patients with CVD are considered to have a higher risk for PAH, which may occur in isolation or in combination with ILD. Clinically, and from a therapeutic perspective, the disease in patients with CVD is indistinguishable from its primary form.
PAH is more common in patients with PSS (10%–33%) and MCTD, especially those with predominant scleroderma features. It is less common in SLE (5%–10%) and even more rare in patients with RA, PM/DM, or SS. The limited cutaneous form of systemic sclerosis, also known as CREST syndrome ( c alcinosis, R aynaud disease, e sophageal dysfunction, s clerodactyly, and t elangiectasis), is very often associated with PAH.
Although the pathophysiologic mechanisms of the disease in CVD are not completely understood, its histologic features resemble those seen in primary pulmonary hypertension. Thromboembolic disease may also play a role in patients with SLE and antiphospholipid antibodies.
The most common manifestation of PAH is dyspnea during physical exertion, a nonspecific symptom that makes it difficult to raise the suspicion clinically. For this reason, it is recommended that all patients with PSS and those with MCTD and PSS features undergo noninvasive screening for the early detection of PAH, usually with transthoracic echocardiography. Right heart catheterization is the reference standard for a definitive diagnosis and should be performed when the diagnosis of PAH is suspected.
CT Signs Indicating Pulmonary Arterial Hypertension
Chest CT in patients with PAH may show an increased diameter of the pulmonary arterial trunk (>30 mm), the main pulmonary arteries and their segments, and, in more advanced cases, dilation of the right heart chambers, with contiguous reflux of contrast into the inferior vena cava and hepatic veins. Associated pericardial effusion is a finding that portends a poor prognosis. Pericardial effusion is more common in patients with severe PAH and may be a manifestation of impaired venous and lymphatic drainage resulting from elevated right atrial pressure. CT is also useful for excluding the presence of pulmonary embolism and assessing concomitant ILD.
Cardiac MRI is a complementary method for diagnosing PAH and is currently considered the reference standard for the noninvasive evaluation of right ventricular structure and function. It also provides better evaluation of pulmonary artery flow and compliance as compared with other imaging methods.
Collagen Vascular Diseases
Rheumatoid Arthritis
RA is a chronic inflammatory disease, typically involving the small joints of the hands and feet in a symmetric fashion. The disease affects mainly women, with peak incidence between 25 and 50 years. Extraarticular manifestations of RA are frequent and may occur in virtually all organ systems. Smoking is associated with a higher risk of developing RA and also with severe forms of the disease.
Main Thoracic Findings
Although RA affects mainly women, pulmonary diseases are more common in men. Respiratory manifestations usually become more prevalent as RA progresses but may present simultaneously with joint symptoms or even predate joint involvement. The prevalence of the different types of RA-associated respiratory diseases is difficult to estimate, but it appears that ILD and pleural disease are most common and ILD, constrictive bronchiolitis, drug reactions, and infections have the greatest impact on patient outcomes.
Most Common Interstitial Findings
The reported prevalence of RA-associated ILD varies significantly, depending on the method of detection and the population examined. ILD had a prevalence initially reported to be around 1.6% to 5%. With the use of HRCT, the prevalence of ILD in unselected RA patients has been found to be as high as 63%. However, clinically significant disease is less common and estimated to occur in approximately 10% of patients.
The main histologic patterns of ILD associated with RA are UIP ( Fig. 23.2 ), NSIP, organizing pneumonia (OP), and diffuse alveolar damage (DAD). Although the radiographic and histologic patterns of NSIP are the most common in the connective tissue disease as a whole, UIP is the most common pattern in RA patients.


Other Thoracic Findings
Pleural involvement is the most common manifestation of thoracic disease in RA, with a frequency of 4% to 75% in postmortem studies. Pleural effusions frequently occur during periods of active arthritis and in patients with subcutaneous nodules, mostly men in their 40s and 50s.
Pulmonary rheumatoid nodules are more common in men than in women and usually result in no symptoms. They are frequently located in the periphery of the right middle lobe or in the upper lobes, ranging from millimeters to several centimeters in diameter. Central cavitation occurs in about 50% of the nodules. Calcification is rare in contrast to silicotic nodules, which commonly calcify. Pulmonary nodules are usually found in patients who have subcutaneous nodules and positive rheumatoid factor.
Progressive Systemic Sclerosis
PSS, or scleroderma, is a chronic multisystem disorder of small vessels and connective tissues characterized by diffuse fibrosis of the skin and internal organs, most frequently the lungs and gastrointestinal tract. The disease has a 3 : 1 female predilection and typically occurs in the third to fifth decades of life. Almost all patients have skin involvement. Patients with ILD and pulmonary vascular disease may be severely symptomatic due to restrictive lung physiology and low diffusing capacity. Esophageal dysmotility occurs in almost all PSS patients, and gastroesophageal reflux may mimic or overlap with ILD. Cardiac involvement, including right heart dysfunction due to PAH, myocardial fibrosis, and pericardial involvement, may further compound pulmonary symptoms.
Main Thoracic Findings
ILD and PAH are the most common cardiopulmonary findings in PSS. About two-thirds of patients suffering from PSS develop ILD. PAH is present in about 20% of PSS patients and is typically associated with severe pulmonary disease, although it may be an isolated manifestation of PSS. Currently, ILD is the leading cause of death in PSS patients, due to pulmonary fibrosis with or without PAH. The mortality rate from ILD is about 40% within 10 years after the onset of the disease.
Most Common Interstitial Findings
Among interstitial pneumonias, the most frequently seen in PSS patients is NSIP ( Fig. 23.3 ). Typically, the disease affects the posterior regions of the lower lobes, with initially subtle ground-glass opacities, as well as accentuated reticulation that may progress to frank pulmonary fibrosis; this is defined as architectural distortion with reticulation, traction bronchiectasis and bronchiolectasis, and honeycombing.