The complexity of the congenital anomalies of the spine can make the neuroradiologic diagnosis challenging. Knowledge of spinal embryology greatly helps in the understanding and classification of these anomalies. We use the classification devised by Tortori-Donati and Rossi and find it helpful from clinical and imaging standpoints. We believe that most patients who have known or suspected congenital spinal anomalies benefit from MR imaging.
The spine is a complex anatomic structure that is formed by elaborate mechanisms. Congenital spinal anomalies tend to be complex, and their names and classification are often confusing. Although most congenital spinal anomalies present in the newborn period, some of them (particularly the so-called “occult” or “closed” ones) may not be found until adulthood. Severe or “open” dysraphisms and those also affecting the development of the distal gastrointestinal and genitourinary tracts and of the lower extremities are generally rapidly diagnosed and may require immediate treatment to avoid complications. At least a basic understanding of the development of the spine is needed before attempting to interpret imaging studies of congenital spinal anomalies. We begin this article with a brief overview of spine development because discreet anomalies can generally be traced back to failure of specific embryologic mechanisms. We attempt to follow the classifications of these anomalies proposed by Tortori-Donati and Rossi but have taken the liberty to insert our own points of view. Regardless of our opinion, the classification developed by Tortori-Donati and Rossi is a valid and valuable one. We believe that most congenital spinal anomalies are better portrayed with MR imaging than with any other imaging technique; therefore, the use of MR imaging is emphasized throughout this article. We address the more commonly encountered congenital anomalies involving the spine, with emphasis on dysraphisms and masses.
Embryology
During the first stage of embryologic development (called the “pre-neurulation” stage), the primitive streak forms on approximately day 17 and consists of a longitudinal primitive groove and a primitive node (Hensen’s node) with a central pit at the presumptive cranial end ( Fig. 1 ). These structures are located on the dorsal (chorionic) surface of the bilaminar embryonic disc. At this time, there is an invagination of cells near the primitive streak between the epiblast (future ectoderm) and the endoderm, forming the mesoderm. This process is called gastrulation. The mesoderm cells that travel through the primitive pit and migrate cranially form the notochordal process (the notochord process initially is a hollow tube called the “notochordal canal”), which parallels the streak. The ventral aspect of the notochordal process fuses to the subjacent endoderm and opens ventrally from the region of the primitive pit, effectively unzipping itself and allowing for a transient communication between the yolk sac and amniotic (ventral surface) cavities. Anomalies at this stage include the so-called “split notochord” anomolies, of which the most common is diastematomyelia. The least common anomolies are intraspinal enteric cysts and fistulas. The open neural tube then flattens and is contiguous with the embryonic endoderm as the notochordal plate. At approximately day 23, the notochordal plate detaches from the neural tube (embryonic ectoderm) and is converted in the process to the definitive solid structure known as the notochord (by obliteration of the notochordal cavity) ( Fig. 2 ).

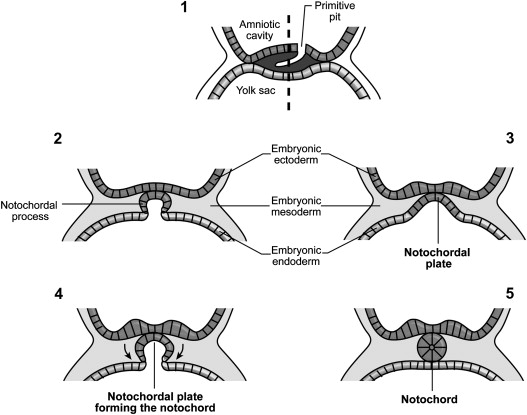
The notochord does not form elements of the spinal column; however, it plays an important role in the induction of the vertebral bodies (via the para-axial mesoderm) and of the neural tube. Likely in response to induction from the underlying notochordal plate and prechordal plate, a focal area of thickening appears in the surface of the epiblast, called the neural plate. While the cranial portion of the neural plate forms the brain, the caudal portion, overlying the notochord, becomes the spinal cord.
The second stage of development is called “primary neurulation” (from day 17 to about day 28 of life) and is responsible for the formation of nearly 90% of the spinal cord. The caudal portion of the neural plate elongates rapidly from days 22 to 26, and there is a respective elongation of the underlying notochord. During this period, a groove forms along the central portion of the neural plate, and its lateral margins become thickened and raised. These thickened lateral margins are called the “neural crests.” At these levels, a group of specialized cells (the neural crest cells) originate and later migrate ventrally to establish peripheral nerves and various mesodermal and vascular structures. The neural crests continue to fold in a concave fashion to meet in the midline and eventually close to form a tube-like structure (the “neural tube”). An increased gradient of bone morphogenic protein establishes the dorsal surface of the neural tube, while an increased gradient of sonic hedgehog gene locates the ventral aspect of the neural tube (therefore establishing the posterior and anterior surfaces of the spinal cord). The spine is established by the presence of repetitive segments called rhombomeres. The organization of these rhombomeres is highly preserved from lesser animals to human beings. These rhombomeres express a variety of genes whose presence aids in the organized development of the spine. Traditionally, the neural tube has been assumed to first fuse in its center and then to extend this closure cephalad and caudally in a zipper-like fashion. This mechanism fails to explain the presence of open spinal dysraphisms in sites other then the extremes of the neural tube. Thus, it is likely that neural tube closure occurs simultaneously at several sites also in a zipper-like manner. From this explanation, it is obvious that failure of primary neurulation results in “open” dysraphisms, which are a group of disorders in which a portion of the neural tube remains exposed and visible (the most common of these disorders is the myelomeningocele).
Once closure of the neural tube commences, it detaches itself from the overlying ectoderm in a process called “disjunction.” Disjunction may occur before its appropriate time (so-called “premature disjunction”) or late (so-called “incomplete disjunction”). A variety of disorders may arise from either of these two abnormal mechanisms.
The third and last stage in the development of the spine is called “secondary neurulation” and involves the formation of about 10% of the spine, all of it in the caudal-most region. In this process, the most important mechanism is that of “canalization and retrogressive differentiation.” The distal-most notochord and neural tube merge inferiorly at the caudal cell mass. The undifferentiated cells in this mass give rise to the bones of the sacrum, coccyx, and probably the fifth lumbar vertebra. They induce cavitation of the distal neural tube until it contains only one central canal (canalization) ( Fig. 3 ). This canalized tube regresses in a cephalad manner (ie, retrogressive differentiation), establishing the nerve roots and the conus medullaris. Complete regression of its cells leaves behind a strand that is made off only pia and ependyma (the filum terminale). Incomplete closure of the central canal in the conus medullaris results in the presence of a terminal ventricle. Exaggerated retrogressive differentiation may be one cause responsible for the caudal regression (or agenesis) syndrome. Maldifferentiation of the caudal cell mass may also contribute to the development of sacro-coccygeal teratomas.

Pathology
Developmental errors can occur at any stage of embryogenesis, alone or in tandem. This can create confusion when making a neuroradiologic diagnosis. Rossi and Tortori-Donati and colleagues have written extensively on the classification of spinal dysraphisms, attempting to simplify this task and remove ambiguity . They suggest that the initial classification should be made clinically by determining whether there is an open or closed spinal dysraphism (ie, whether the defect is open to the air, thus leaving the neural elements exposed and visible) or is covered by a layer of skin, which would prevent the visualization of the neural elements . If closed, the lesion can be further characterized by whether or not there is an associated mass. Clues as to the presence of an underlying closed dysraphisms may be found during the physical examination and include a midline dimple, dermal sinus, port wine stain, focal hirsutism, lower extremity deformities, and anal and genitourinary anomalies . Spinal dysraphisms are often diagnosed prenatally by the presence of elevated maternal alpha fetoprotein in cases of open neural tube defects and by prenatal sonography and MR imaging . In some cases, in utero surgical repair of the defect may be performed .
Open dysraphisms
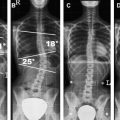
Open spinal dysraphisms include myelomeningoceles, myeloceles, hemimyelomeningoceles, and hemimyeloceles, all of which have associated Chiari II malformations of varying degrees of severity. Myelomeningoceles account for nearly 99% of open spinal dysraphisms. In developing countries, the overall incidence of myelomeningocele is about 2 out of 1000 live births, whereas in industrialized nations and with the administration of preconception and prenatal maternal folates, their incidence has dropped to about 2 out of 10,000 live births. MR imaging and sonography permit for in utero diagnosis of myelomeningoceles ( Fig. 4 ). Myelomeningoceles are usually easily diagnosed clinically by the presence of a large protruding mass containing the nonneurulated neural tissue called a “placode.” In these patients, a prominent ventral subarachnoid cerebral spinal fluid–filled space pushes the placode away from the surface of the skin ( Fig. 5 ). Myeloceles also have an exposed placode; however, there is no associated expansion of the subarachnoid space and the placode remains flush with the cutaneous surface. Both defects are usually found at the lumbosacral level but may occur elsewhere in the spine. Accompanying all open defects is an incomplete migration of the para-axial mesenchyme, resulting in a bony spinal defect (spina bifida) and significant muscle atrophy, both of which contribute to the development of scoliosis. This is the origin of the term “spinal bifida aperta,” which is no longer used. In the rare case of tandem errors in gastrulation and neurulation, there is a diastematomyelia, and one of the hemicords is contained in a myelomeningocele or a myelocele resulting in a hemimyelomeningocele or hemimyelocele (both rare anomalies). MR imaging is the modality of choice to preoperatively assess the components of these malformations, to define the anatomic relationship of the placode with the underlying nerve roots, and to search for associated abnormalities. Many surgeons repair the dysraphisms without obtaining an imaging study of the spine. Some type of imaging study of the brain is needed to exclude the presence of hydrocephalus (found in 75% of patients with open dysraphisms), which may be shunted at the time of the spinal repair.
In open dysraphisms, typically there is a low-lying dysplastic spinal cord, and the placode forms the posterior wall of the defect. In the case of the myelomeningocele, ventral to the placode there is a dilated subarachnoid space, through which the nerve roots traverse. The nerve roots arise from the ventral placode, which is the nontubulated outer surface of the cord. The conus medullaris is not normally formed, and imaging of the entire spine is needed to exclude the presence of a cord cavity (syringohydromyelia) or other defects, such as diastematomyelia.
Although one of the surgical goals is to untether the placode, reform it, and replace it in the native spinal canal, most placodes remain tethered to the site of repair. Therefore, postoperative imaging is performed to search not only for the status of the placode but also for other complications, such as developing hydromyelia, constriction dural rings at the site of grafting and repair, epidermoids and dermoids, ischemia of the cord, and diastematomyelia .
Closed dysraphisms with associated mass
We begin this discussion by addressing closed dysraphic states that have associated back masses. These include lipomyeloceles, lipomyelomeningoceles, meningoceles, and myelocystoceles. Lipomyeloceles and lipomyelomeningoceles also occur secondary to abnormal neurulation, where dysjunction occurs prematurely and allows primitive mesenchymal cells to migrate into the neural tube. When these cells reach the placode, they are induced to form adipose tissues. The result is the formation of a lipoma that traverses the bifid bony posterior elements and is contiguous with the epidural and subcutaneous fat. If the placode–lipoma complex remains within the spinal canal, the abnormality is called a lipomeningocele (analogous to the meningocele); if it lies outside of the confines of the spinal canal because the ventral subarachnoid space is expanded, it is called a lipomyelomeningocele (analogous to the myelomeningocele) ( Figs. 6 and 7 ) .
A meningocele is a rare anomaly that manifests as a herniation of meninges through a neural foramen laterally or through a vertebral or sacral defect anteriorly or posteriorly . The herniated sac consists of an outer layer of dura and an inner arachnoid membrane and usually contains only CSF, although occasionally it may contain nerve roots, dysplastic tissues, or tumor . Often, meningoceles are associated with systemic mesenchymal disorders, such as neurofibromatosis type 1 or Marfan syndrome, but can be seen in isolated forms ( Fig. 8 ) . Over 80% of meningoceles are posterior and in the lumbosacral spine, but they are occasionally found in the thoracic spine and sacrum and may become large enough to cause symptoms from mass effect ( Fig. 9 ) . Spinal meningoceles are thought to be caused by failed closure of the neural tube in the neurulation stage of development and then gradually increase in size secondary to continual CSF pulsations . Traditionally, these lesions were evaluated with plain radiographs, CT, and myelography, but MR imaging is now used most frequently because it allows for better characterization regarding its anatomic relationships and contents and may reveal other associated congenital lesions, such as genitourinary anomalies or spinal anomalies . Eroded vertebral bodies, thinned vertebral arches, and expanded foramina are associated findings . Spinal sonography in the newborn may show a cystic mass in an expanded spinal canal, with displacement of the spinal cord by the meningocele .
A myelocystocele is a large posterior meningocele that lies dorsal to a bony spina bifida and houses a large terminal syrinx (syringocele). The meningocele component is continuous with the subarachnoid space, and the syringocele is continuous with the ependymal canal. There is no communication between the two spaces . This anomaly is rare.
Closed dysraphisms without associated mass
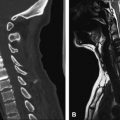
Closed dysraphisms without an associated mass include intradural, intramedullary, or filar lipomas and the tight filum terminale syndrome. Intradural and intramedullary lipomas lie within the dural sac, most commonly in the lumbosacral region, although they may be found at any level ( Fig. 10 A). At the level where they are contiguous with spinal cord, there is a placode, which often displaces the spinal cord away from the midline ( Figs. 10 B and 11 ). On MR imaging, lipomas should follow the signal intensity of subcutaneous fat in all sequences. Hyperintense T1 signal in a thickened filum terminale indicates a filar lipoma, which may be considered incidental if the patient does not have clinical signs of tethered cord syndrome and if the conus medullaris terminates at a normal level (never below the L2-L3 disc space) ( Fig. 12 ). Filar lipomas are probably due to an anomaly of secondary neurulation (although some authors think that they are due to anomalies of retrogressive differentiation). The tight filum terminale syndrome develops due to abnormal differentiation of the secondary neural tube, causing the filum to be shortened and hindering the apparent ascent of the conus medullaris . The filum can be considered to be thick when it measures more than 2 mm in diameter or is significantly thicker than the adjacent nerve roots. Symptomatic patients who have a thick filum and an abnormally low conus medullaris benefit from surgery.
Around the third week of gestation, when the cells from Hensen’s node are proliferating and migrating, there may be a focal splitting of the notochord secondary to a persistent connection (canals of Kovalesky) between the endoderm and ectoderm . Although there is debate over the exact mechanism, failure of midline integration of the notochord anlagen may result in a spectrum of rare anomalies with variable complexity. A dorsal enteric fistula is considered to be the most severe form, resulting in a persistent connection between the bowel and the posterior cutaneous surface, crossing between a duplicated spine . Displaced endodermal tissue may be located anywhere along the path of the abnormal connection, resulting in a neurenteric cyst, which may occur at any level of the spine but most commonly occurs in the lower cervical and upper thoracic regions. Neurenteric cysts may be intraspinal, intramedullary, or postvertebral but are often are prevertebral, located in the posterior mediastinum . These cysts are lined by alimentary or respiratory epithelium and may produce mucus. Patients may present with signs of spinal cord compression from mass effect of the cyst or with meningitis in cases when the cyst ruptures into the subarachnoid space . Multiple associated abnormalities have been described, including vertebral/spinal and cardiac anomalies, renal and limb dysgenesis, and various anomalies of the alimentary tract, such as partial duplications and fistulas . “Split notochord syndrome” refers to associated vertebral anomalies, central nervous system abnormalities, and intestinal anomalies . The imaging appearance is variable, depending on the location and severity of the abnormality. Depending on the protein content of the cystic material, the fluid may be iso- to hyperintense on T1- and T2-weighted images . These lesions generally do not enhance . The sine qua non for a neurenteric cyst is a wide anterior spina bifida through which there is a direct communication between a cystic posterior mediastinal mass and the subarachnoid space .
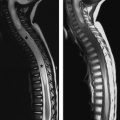
If the notochord fails to fuse in the midline because of a persistent connection between the yolk sac and amniotic cavities and if there are cells from the primitive streak between the anlagen, two separate hemicords are formed. This is referred to as “diastematomyelia.” The final outcome is dependent on progression or regression of the cells from the primitive streak. With progression, two separate neural plates are formed, and the hemicords eventually lie within two separate dural tubes, divided by a thick osteocartilaginous septum. This is called a “type I diastematomyelia” and is symptomatic due to tethering of the cord by the spur and often the presence of syringomyelia ( Fig. 13 ). “Type II diastematomyelia” occurs when the cells of the primitive streak regress, with the end outcome being a single dural tube housing both hemicords, which may or may not be separated by a thin, nonrigid fibrous septum . In the absence of a septum, these anomalies are nearly always asymptomatic but may come to clinical attention if a syrinx develops ( Fig. 14 ).
Thought to be a result of focal areas of incomplete disjunction, dermal sinuses are tracts lined by squamous epithelium that connect the skin to deeper tissue layers and may extend to the intramedullary spinal cord space . They occur in the dorsal midline at any level of the spine but are most commonly are found in the lumbosacral region . The tracts may contain dermal and/or neuroglial elements and may traverse several levels in their course into the subarachnoid space, depending on the point at which the sinus was formed and how far the spinal cord ascended within the spinal canal from that point of development . Patients who have dermal sinuses often have a skin dimple or a cutaneous pit in the midline of their back on physical examination. Other associated abnormalities include a tethered cord, (lipo) myelomeningocele, and vertebral body anomalies . The tracts can be seen on cross-sectional imaging studies, such as MR imaging or sonography . MR imaging may be helpful because it allows for visualization of the entire course of the sinus tract and the associated pathology(eg, skeletal abnormalities, spinal cord abnormalities such as syringohydromyelia, and masses such as dermoids and epidermoids) . The sinus tract may enhance if infected, and abscess may occur along its tract.
Spinal dermoid and epidermoid cysts (or nodules) are thought to develop secondary to ectodermal tissue remaining within the neural tube after primary neurulation . These lesions are often associated with dermal sinus tracts but may also be found in isolation . Histologically, epidermoid cysts have an outer wall of stratified squamous epithelium surrounded by a collagenous layer and a central cystic component created by desquamation and keratin breakdown, which may contain cholesterol crystals and fatty acids. Calcium may be present in the wall or within the cavity . Dermoid cysts are similar but also contain skin appendages (ie, glandular elements and hair follicles) . Both may be found in extradural, subdural, or intramedullary locations . Epidermoid cysts are found most commonly in the thoracic spine between the levels of T5 and T8 . Dermoid cysts are most commonly located in the lumbosacral spine but have also been reported in the cervical and thoracic spine . Both may be seen in patients who have the Klippel-Feil syndrome. The findings on MR imaging are somewhat variable for dermoids and epidermoids. Homogeneous hypointense T1 and hyperintense T2 signals are typically seen, but heterogeneous signal intensity with patchy areas of enhancement has also been reported ( Fig. 15 A) . Enhancement generally indicates current or previous infection. Diffusion-weighted imaging has been shown to be useful in the diagnosis of spinal epidermoid cysts, showing restricted diffusion within the lesion and thus allowing differentiation from other cystic masses ( Fig. 15 B) .
Spinal hamartomas are benign abnormally located rests of well differentiated mature ectodermal and mesodermal elements . Their etiology is not known, but it has been postulated that it may be similar to intraspinal lipomas, where dorsal mesenchyme is able to enter the neural tube secondary to premature disjunction and is subsequently induced to form these hamartomatous elements by the neural tube . Spinal hamartomas may be located in the cervical, thoracic, or lumbar spine and in the sacrum . A true hamartoma of the spine or of the spinal cord contains only local elements (eg, nerves, blood vessels, fat, bone, cartilage, or synovial membranes), but other elements foreign to this location (eg, lymphoid tissue, glandular tissue, and urinary tract tissues) may be present . Patients who have spinal hamartomas may have no external abnormalities, but there may be a skin dimple or angioma in the overlying skin or a subcutaneous mass. The masses are usually isointense to the spinal cord on T1- and T2-weighted MR imaging, and there may be associated widening of the spinal canal . Symptoms may be due to compression of the spinal cord by the mass.
When all three germ cell layers are represented in a mass, the resulting tumor is called a “teratoma” (also referred to as a “monster neoplasm”) . Sacrococcygeal teratomas are the most common congenital tumors, probably arising from totipotential cells of Henson’s node . They are often associated with other abnormalities, including anorectal and genital malformations, ventricular septal defect, hip dislocation, and vertebral anomalies, such as spina bifida and sacral agenesis ( Fig. 16 ) . Several classifications exist for these tumors. Histologically, they are classified as mature or immature and graded from 0 to 3, with the grade increasing with the amount of immature tissue found in the tumor. Grade 0tumors have only mature tissues, whereas Grade 3 tumors have large amounts of immature tissue . There is also a classification system used by the American Academy of Pediatrics’ surgical section, which grades tumors by their anatomic extent (type I–IV). A type I tumor is mainly external, with a small presacral component; type II is characterized by external presentation with a large intrapelvic extension; type III is characterized by an external component with the majority of the tumor residing in the pelvis and abdomen; and type IV is entirely presacral, without an externally visible component . Teratomas may be cystic, solid, or mixed, and more than 50% contain calcium . These tumors are often first demonstrated on prenatal sonograms and are associated with polyhydramnios . CT and MR imaging show a heterogeneous, enhancing mass with macroscopic fat; cystic areas and calcification are often seen. Although benign tumors are more likely to be predominantly cystic, imaging characteristics cannot reliably predict their histologic grade . MR imaging is ideal for evaluating the intraspinal extension of these teratomas.