▪
Introduction
Congenital heart and vascular diseases are relatively uncommon malformations of the heart or great vessels that occur during embryologic development. With advances in medical and surgical treatments, most patients survive into adulthood, and it is imperative for the chest radiologist to be familiar with the imaging findings of common malformations and their postoperative appearance. Although echocardiography remains the first step in the assessment of these patients, these imaging modalities, with technical advances in CT and MRI, have become key elements in the evaluation of the anatomy and detection of the defects and functional assessment of patients with congenital heart and vascular abnormalities.
▪
Imaging Techniques
Both electrocardiographic (ECG)-gated CT and MRI play important roles in the detection of congenital anomalies of the heart and great vessels.
Electrocardiographic-Gated CT
This is an imaging modality with great spatial resolution and is therefore a useful tool to assess congenital anatomic defects, associated abnormalities, postoperative anatomy, and complications in the postoperative patient. Each examination should be tailored to answer the clinical question posed. Unenhanced, low-dose images are typically useful to identify high-density postoperative changes and avoid misinterpretation of the contrast-enhanced images. The contrast injection protocol, contrast volume, and/or contrast mix should be determined specifically for each patient. With the use of ECG gating, coronary artery analysis can be performed. Although cardiac MRI is more accurate in the evaluation of ventricular volumes and function, ventricular volumes and function can also be quantified using dedicated postprocessing software with the use of retrospective gating CT imaging. The use of delayed imaging can be helpful when trying to assess patency of baffles and/or conduits, especially in patients with Fontan morphology, due to their delayed filling.
Cardiac MRI
Cardiac MRI allows evaluation of the anatomy and functionality of the heart. It is the gold standard for the evaluation of ventricular volumes and function. Every cardiac MRI examination is tailored to answer the clinical question presented. The use of noncine, black blood, and bright blood sequences through the chest are useful in the assessment of the mediastinum and great vessels. The use of bright blood cine images is useful in the assessment of the anatomy and functionality of the heart. Phase contrast images are performed to quantify flow and velocities through a particular imaging plane. Phase contrast images are prescribed orthogonal to the blood flow being assessed to obtain the most accurate evaluation. Phase contrast images allow reproducible flow and velocity quantification and are commonly used during valvular assessment ( Fig. 28.1 ). In patients with congenital heart disease, this imaging technique becomes paramount for the evaluation of intracardiac shunts by quantifying Qp:Qs and by quantifying valvular function. MRI angiography is often used in the assessment of vascular anatomy and associated abnormalities.

▪
General Approach
It is important to have a systematic review of patients with complex congenital heart disease to ensure that all relevant abnormalities are detected and the report is complete. Analysis of the chest should start with assessment of the aortic arch (left or right) and branching pattern. The most common anomalies of the arch are a right-sided arch with mirror image branching pattern (highly associated with congenital heart defects) and a right-sided aortic arch with an aberrant left subclavian artery. It is also important to evaluate the aorta caliber to exclude aneurysm formation as well as detect areas of narrowing, in particular at the aortic isthmus, as can be seen in coarctation. Evaluation of the pulmonary arteries should follow to assess for enlargement of the main pulmonary artery (PA), which is commonly seen in patients with elevated pulmonary arterial pressures. Evaluation of the pulmonary branch arteries can reveal areas of stenosis. Venous connections should be assessed for normal drainage patterns. The most common venous anomaly is a persistent left superior vena cava (SVC). This vein normally courses to the left of the aortic arch and drains into the coronary sinus. Rarely, this vein drains into the left atrium, creating a right-to-left shunt. It is important to review the connections of the pulmonary veins into the left atrium to exclude anomalous pulmonary venous return. When evaluating the heart, it is important to remember the segmental approach developed by Van Praagh et al. based on embryologic development. It consists of three steps. The first is to determine the visceroatrial situs; the second consists of determining the morphologic right versus left ventricle and their relation to each other; and the third consists of defining the ventriculoarterial (VA) connections and their relation to each other.
The orientation of the base-apex of the heart should be described separately—dextrocardia, mesocardia, and levocardia. The position of the heart within the chest should also be described separately (levoposition, dextroposition, mesoposition). Situs ambiguous, situs inversus with levocardia, and situs solitus with dextrocardia are usually associated with congenital heart defects.
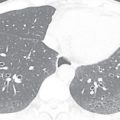
The first step is to determine the visceroatrial situs, which is defined as the relationship between the atria and thoracoabdominal organs. To determine which of the two atria is the morphologic left or right atrium, look at the shape of the atrial appendage. The left atrial appendage has an elongated, finger-like shape; the right atrial appendage has a broad base attachment and triangular shape ( Fig. 28.2 ). Another clue in deciding the left versus the right atrium is the venoatrial concordance. The right atrium is the chamber receiving the inferior vena cava (IVC). When neither of these two methods works to determine the morphologic left versus right atrium, the right atrium should be considered the atrium on the side of the trilobed lung. Once you have determined the left versus right atrium, you now have to determine the position of the liver, spleen, and stomach. There are three types of situs—situs solitus, which is the normal arrangement, situs inversus, which the opposite of the normal arrangement, and situs ambiguous, which is the in-between arrangement. The situs ambiguous is also known as the heterotaxy syndrome and can be further classified into bilateral right sidedness (asplenia) and bilateral left sidedness (polysplenia).

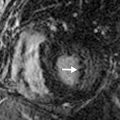
The second step consists of determining the ventricular loop orientation. To determine the rotation of the ventricle, you first have to identify the morphologic right ventricle. The most helpful way to recognize the right ventricle is to identify a muscular infundibulum and resultant muscular separation between the atrioventricular (AV) and semilunar valves ( Fig. 28.3 ). Other important characteristics of the morphologic right ventricle include the presence of a septal leaflet in the AV valve, the presence of a moderator band, papillary muscles that attach to the septum and free wall, and interventricular septal trabeculation. A D-looped right ventricle is the normal looping of the heart, with the right ventricle located on the right position and the aortic valve located to the left of the pulmonic valve. An L-looped right ventricle will have the morphologic right ventricle in the left position and the aortic valve to the left of the pulmonic valve.

The third step includes identifying the position and origin of the great vessels. The normal position of the great vessels is with the PA origin located anterior and to the left of the ascending aorta. Situs inversus in the opposite. In D-transposition, the aorta is located anteriorly and arises from the right ventricle. In L-transposition, there is an anteriorly located aorta arising from the morphologic right ventricle, which is located on the left and is connected to the left atrium. This results in the morphologic right ventricle to the left of the pulmonic valve. The term D and L malposition refers to a normal origin but abnormal position of the great vessels in the chest.
▪
Bicuspid Aortic Valve
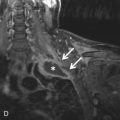
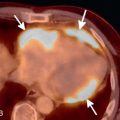
A bicuspid aortic valve is one of the most common types of congenital heart disease, with an estimated incidence of 0.9% to 2%. It is more common in males than in females, with a 2 to 3 : 1 ratio. It is inherited as an autosomal dominant trait with incomplete penetrance and results in incomplete separation of the valve leaflets during development. The most common leaflets to demonstrate the incomplete separation are the left and right cusps (70%–86%), followed by the right and noncoronary cusp (12%). It is imperative to assess the aortic valve morphology during systole because a raphe (fibrous ridge at the site of fusion of the conjoined cusps) can simulate a trileaflet valve during diastole. Bicuspid aortic valves can be associated with aortic coarctation, supravalvular aortic stenosis, subvalvular aortic stenosis, ventricular septal defects (VSDs), patent ductus arteriosus (PDA), and sinus of Valsalva aneurysms ( Fig. 28.4 ). Patients with a bicuspid aortic valve develop aortic stenosis more commonly than patients with a trileaflet valve. To a lesser degree, these patients can also develop aortic regurgitation. Bicuspid aortic valve is considered a risk for infective endocarditis and is often associated with aortic root or ascending aortic dilation. Associated syndromes include Loeys-Dietz and Turner syndromes.

Echocardiography is the most common imaging modality to assess and follow up patients with a bicuspid aortic valve. Its high temporal and spatial resolution, ability to quantify velocities, gradients, and valve areas, as well as the lack of use of ionizing radiation and nephrotoxic contrast agents, make it the imaging modality of choice.
The use of CT and MRI has been growing, in particular in the preoperative evaluation of the aorta and in the follow-up of a thoracic aortic aneurysm. CT, with the use of multiplanar reformats, is a very accurate and reproducible method of measuring the aorta. The use of a retrospective ECG-gated acquisition allows for the morphologic assessment of the valve and planimetry of the valve opening area and/or regurgitant orifice. Cardiac MRI allows for anatomic assessment similar to CT and, with the use of phase contrast images, allows quantification of peak velocities to determine the peak systolic gradient (using the modified Bernoulli equation: gradient = 4v 2 , where v = velocity) to determine the severity of aortic stenosis and flow quantification and the degree of aortic regurgitation.
The type of surgical procedure in a patient with a bicuspid aortic valve depends on whether the patient needs aortic valve intervention, aortic repair, or both. Aortic valve intervention without repair of the aorta can be performed by aortic valvuloplasty, surgical aortic valve replacement, or the Ross procedure. Transcatheter aortic valve replacement has an uncertain role in patients with a bicuspid aortic valve due to the lack of experience. Aortic valvuloplasty or balloon valvotomy is not recommended in older patients with calcified leaflets. It has a role in adolescents and young adults with aortic stenosis without significant calcification. Surgical intervention in adolescents or young adults with symptomatic bicuspid aortic valve is usually accomplished with a bioprosthetic valve. The use of a pulmonary autograft and right-sided reconstruction with a pulmonary homograft (Ross procedure) is limited due to its technical complexity and complications in the aortic autograft, the pulmonic homograft, and the availability of simpler and more effective alternatives (e.g., mechanical and bioprostheses, including stentless bioprosthetic valves).
Combined aortic valve replacement and ascending aorta repair can be done with a prosthetic aortic valve and supracoronary aortic graft. In patients who need aortic valve replacement and have a dilated aortic root and ascending aorta, a composite graft that includes the aortic valve, aortic root, and ascending aorta can be used (Bentall procedure). Re-implantation of the coronaries is needed in this type of procedure. Surgery to replace the aortic root or ascending aorta in patients with a bicuspid aortic valve is performed when the aortic root or ascending aorta measures 5.5 cm or greater. If there is a family history of aortic dissection or rapid growth (>5 mm/year), surgery is suggested at 5.1 to 5.5 cm. If the patient is undergoing aortic valve surgery due to severe stenosis or regurgitation, the aorta should be repaired if larger than 4.5 cm. Repair of the aortic bicuspid valve depends on the patient’s symptoms.
▪
Aortic Coarctation
Aortic coarctation is a congenital narrowing of the descending thoracic aorta, typically at the level of the ductus arteriosus (DA). It accounts for approximately 4% to 6% of all congenital heart defects and occurs more in males than in females. The cause is unknown, but underdevelopment of the aortic arch secondary to decreased anterograde flow and extension of ductal tissue into the wall of the aorta are the two leading theories. A genetic predisposition is present, given the association with Turner syndrome.
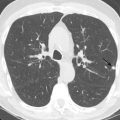
Aortic coarctation is usually seen in conjunction with other congenital abnormalities, including bicuspid aortic valve, single-ventricle variants, endocardial cushion defects, D-transposition, VSDs, atrial septal defects (ASDs), PDA, and aortic or subaortic stenosis ( Fig. 28.5 ). Close association with an aortic aneurysm is also common.

The clinical presentation varies depending on the severity of the narrowing. In cases of severe narrowing, patients present in the neonatal age with heart failure due to increased left ventricular (LV) afterload once the PDA closes. In cases of mild narrowing, patients can go unrecognized until they are adults. In these patients, systolic hypertension recorded in the upper extremities, with low blood pressure in the lower extremities and diminished or delayed femoral pulses, is the classic clinical presentation.
There is a spectrum of anatomic variants, from the classic short-segment coarctation that includes long segmental narrowing, tubular hypoplasia, and pseudocoarctation. ECG-gated CT is a helpful imaging tool for the anatomic assessment of aortic coarctation before and after treatment. With CT’s great spatial resolution, the area of narrowing can be accurately measured. Furthermore, with the visualization of the entire chest during the same image acquisition, you can assess for the presence of collateral vascularity. CT is commonly used as a diagnostic tool and for the follow-up of patients after surgical correction. Cardiac MRI and MR angiography (MRA) are also helpful for the evaluation of these patients and do not use ionizing radiation. With the use of phase contrast images, prescribed at and a few centimeters distal to the coarctation, flow and velocity across the coarctation can be quantified. Using the peak velocity and modified Bernoulli equation (gradient = 4v 2 ), the peak gradient across the coarctation can be estimated. Furthermore, with the prescription of phase contrast images of the distal thoracic aorta (at the level of the diaphragm), you can assess for recruitment of blood flow through collateral vascularity. Any increase in blood flow in the distal descending thoracic aorta relative to the flow at or just distal to the coarctation is considered abnormal and is suggestive of collateral vascularity.
Treatment of aortic coarctation should be performed as early as possible. The type of treatment used (balloon angioplasty ± stenting vs. surgery) depends on the expertise of the treating team and associated malformations.
Problem Solving
Trying to differentiate between coarctation and pseudocoarctation can be difficult. The imaging characteristics that can differentiate them are as follows.
An elongated aorta anchored at the ligament arteriosus is compatible with a pseudocoarctation. It is also called kinking or buckling of the aorta . No significant narrowing is noted in pseudocoarctation with gradients less than 20 mm Hg on MRI phase contrast images. No collateral vascularity is present with pseudocoarctation.
▪
Atrial Septal Defects
ASDs account for approximately 13% of congenital heart disorders. ASDs can be classified as defects resulting from abnormal development of the interatrial septum (e.g., in ostium secundum and ostium primum defects) and defects resulting in interatrial communication with a normally developed interatrial septum (e.g., sinus venosus and unroofed coronary sinus defects).
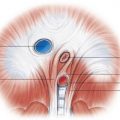
It is important to discuss the embryologic development of the interatrial septum briefly to understand ASDs and their imaging appearance better. The normal septation of the atria begins in the fifth week of gestation, with the septum primum growing from the superior aspect of the common atria down toward the endocardial cushions. The gap between the free end of the septum primum and the endocardial cushions is called the ostium primum . The fusion between the septum primum and the endocardial cushions closes the ostium primum. Multiple small fenestrations (that later coalesce) occur in the septum primum, called the ostium secundum . The septum secundum forms as an invagination of the atrial wall to the right of the septum primum. As the septum secundum continues to grow caudally, it eventually covers the ostium secundum. It is important to know that the septum secundum does not completely divide the two atria, leaving an oval-shaped hole called the fossa ovalis . The fossa ovalis is covered solely by the overlapping septum primum. At birth, with the expansion of the lungs, the right-sided pressures drop, causing a left-to-right gradient, which results in permanent opposition of the septum primum against the septum secundum. In two- thirds of the population, the septum primum fuses permanently with the septum secundum and, in the remaining population, there is a probe-patent fossa ovalis (patent foramen ovale [PFO]; Fig. 28.6 ).

An ostium primum ASD corresponds to 15% to 20% of all ASDs. It results from an abnormal fusion between the septum primum and endocardial cushions, resulting in a persistent ostium primum defect. It is considered the mildest form of an endocardial cushion defect. This type of defect is uncommon and usually is seen as part of an endocardial cushion defect, with an associated VSD and mitral and/or tricuspid valve clefts ( Fig. 28.7A and B ).