Transthoracic echocardiography (TTE) is the primary imaging modality in patients with congenital heart disease. It is portable, affordable, and has excellent spatial and temporal resolution. Transesophageal echocardiography (TEE) can be complementary, but in growing adults with congenital heart disease, ultrasound modalities are often limited by suboptimal acoustic windows. Cardiac magnetic resonance (CMR) imaging is independent of acoustic windows. It is a one-stop shop that allows the evaluation of intra and extracardiac anatomy, quantification of ventricular function and valvar regurgitation shunt quantification, tissue characterization, and stress imaging. It is a long test that requires sedation in children. It is susceptible to metallic artifacts and expertise in the performance and interpretation of congenital is limited. It is also contraindicated with certain pacemakers, defibrillators, or other implanted electronic devices. Cardiac computed tomography (CCT) provides excellent spatial resolution, especially for coronary artery delineation and evaluation of the pulmonary vasculature, the bronchial tree, and lung parenchyma. It is a fast test yet does expose patients to harmful radiation albeit the radiation exposure has decreased with the newer generation technology. CCT has a lower temporal resolution compared to TTE, TEE, and CMR. Invasive cardiac catheterization (ICA) is the modality of choice for interventional procedures and hemodynamic assessment. It is invasive and does expose patients to harmful radiation. Radionuclide myocardial perfusion scintigraphy is the main functional cardiac imaging technique for the assessment of ischemic heart disease in adults. In congenital heart disease, it can be used in high-risk populations who underwent coronary artery implantation. Yet it is rarely used in children and young adolescents because of its high false positive rates as well as exposure to harmful radiation.
A proposed algorithm for the use of multimodality imaging in congenital heart disease is seen in Fig. 12.1 .

Acyanotic Congenital Lesions
Cardiac Shunts
Clinical Overview
Cardiac shunts are the most common congenital heart defects. They are abnormal connections between the cardiac chambers or the great vessels. Hemodynamically significant left to right shunts (Qp/Qs >= 1.5) can cause cardiac chamber enlargement distal to the shunt, pulmonary overcirculation failure to thrive, and heart failure and arrhythmias. When left untreated hemodynamically significant shunts can result in elevated pulmonary vascular resistance and pulmonary hypertension in adulthood.
Atrial septal defects (ASD) and other forms of interatrial communications are common and can occur as a consequence of deficiency in the septum primum (secundum ASD; Figs. 12.2, 12.3–12.5 ,  ), endocardial cushions (primum ASDs; Fig. 12.6 ,
), endocardial cushions (primum ASDs; Fig. 12.6 ,  ), in the wall separating the sinus venosus region from the right-sided pulmonary veins (sinus venosus defects; Fig. 12.7 ,
), in the wall separating the sinus venosus region from the right-sided pulmonary veins (sinus venosus defects; Fig. 12.7 ,  ,,, ), and finally the wall separating the coronary sinus from the left atrium (coronary sinus septal defect; Fig. 12.8 ,
,,, ), and finally the wall separating the coronary sinus from the left atrium (coronary sinus septal defect; Fig. 12.8 ,  , ). Secundum ASDs are the most common. Hemodynamically significant interatrial communications cause right heart enlargement, right ventricular dysfunction, and in a small percentage pulmonary arterial hypertension. Hemodynamically significant interatrial shunts are often closed in childhood by may require surgical or transcatheter interventions. Unrepaired ASDs may present with arrhythmias in adults.
, ). Secundum ASDs are the most common. Hemodynamically significant interatrial communications cause right heart enlargement, right ventricular dysfunction, and in a small percentage pulmonary arterial hypertension. Hemodynamically significant interatrial shunts are often closed in childhood by may require surgical or transcatheter interventions. Unrepaired ASDs may present with arrhythmias in adults.







Isolated ventricular septal defects (VSD) are the most common congenital lesions diagnosed in childhood. They often present with a murmur and when hemodynamically significant result in left-sided heart enlargement, pulmonary overcirculation, and congestive heart failure. They are classified based on their location in the ventricular septum into membranous ( Figs. 12.9, 12.10 ,  , ), inlet, outlet, or conal ( Fig. 12.11 ,
, ), inlet, outlet, or conal ( Fig. 12.11 ,  ), and muscular. Small membranous and muscular defects tend to close spontaneously. Small VSDs have excellent long-term survival, though they can present in adulthood because of double-chambered right ventricle (DCRV), infective endocarditis, aortic valve prolapse, and aortic regurgitation. In adulthood, VSDs are encountered either as small patch leaks after surgical repair, as unrepaired small pressure restrictive defects (i.e., Qp/Qs < 1.5 and normal pulmonary vascular resistance) or, as a hemodynamically significant defect that has developed Eisenmenger syndrome with systemic or suprasystemic pulmonary vascular resistance resulting in shunt reversal and cyanosis.
), and muscular. Small membranous and muscular defects tend to close spontaneously. Small VSDs have excellent long-term survival, though they can present in adulthood because of double-chambered right ventricle (DCRV), infective endocarditis, aortic valve prolapse, and aortic regurgitation. In adulthood, VSDs are encountered either as small patch leaks after surgical repair, as unrepaired small pressure restrictive defects (i.e., Qp/Qs < 1.5 and normal pulmonary vascular resistance) or, as a hemodynamically significant defect that has developed Eisenmenger syndrome with systemic or suprasystemic pulmonary vascular resistance resulting in shunt reversal and cyanosis.



There are different types of atrioventricular septal defects ( AVSD ; partial, transitional, and complete), but overall, the pathophysiology of AVSD is a lack of fusion of the endocardial cushions ( Figs. 12.12 and 12.13 ,  , ), which will initially cause a large left-to-right shunt. Due to this large left-to-right shunt, pulmonary overcirculation and congestive heart failure symptoms are common in the first few months of life. Repair occurs early in childhood, but complications after surgical interventions can be seen into adulthood. Re-operation for atrioventricular valve regurgitation residual shunts or left ventricular outflow tract obstruction may be indicated in adulthood. Arrhythmias, left ventricular dilation or dysfunction may be seen in adulthood. Thus these patients require lifelong follow-up and monitoring.
, ), which will initially cause a large left-to-right shunt. Due to this large left-to-right shunt, pulmonary overcirculation and congestive heart failure symptoms are common in the first few months of life. Repair occurs early in childhood, but complications after surgical interventions can be seen into adulthood. Re-operation for atrioventricular valve regurgitation residual shunts or left ventricular outflow tract obstruction may be indicated in adulthood. Arrhythmias, left ventricular dilation or dysfunction may be seen in adulthood. Thus these patients require lifelong follow-up and monitoring.


Patent ductus arteriosus ( PDA , Figs. 12.14 and 12.15 ,  ) is a vascular connection between the aorta and the main pulmonary artery present in fetal life and that typically closes shortly after birth. It can remain patent in 0.3%–0.8% of term infants and is twice as common in females as males. Like other shunt lesions, the clinical and hemodynamic manifestations of a PDA depend on its size and the systemic and pulmonary vascular resistance. Small PDAs can be inaudible and are not hemodynamically significant while large ones can result in left heart enlargement, congestive heart failure, and eventually lead to pulmonary hypertension. Most hemodynamically significant PDAs are closed in infancy via a transcatheter or surgical-based approach.
) is a vascular connection between the aorta and the main pulmonary artery present in fetal life and that typically closes shortly after birth. It can remain patent in 0.3%–0.8% of term infants and is twice as common in females as males. Like other shunt lesions, the clinical and hemodynamic manifestations of a PDA depend on its size and the systemic and pulmonary vascular resistance. Small PDAs can be inaudible and are not hemodynamically significant while large ones can result in left heart enlargement, congestive heart failure, and eventually lead to pulmonary hypertension. Most hemodynamically significant PDAs are closed in infancy via a transcatheter or surgical-based approach.


Anomalous pulmonary venous connections occur when the pulmonary veins anomalously connect to a systemic vein or the right atrium. Total anomalous pulmonary venous connections (TAPVC) present with cyanosis and are surgically repaired in infancy. They can present as a surgical emergency when the connection to the systemic veins is obstructed. Partial anomalous pulmonary venous connections (PAPVC) present like an ASD with a left to right shunt and the potential for right heart volume overload depending on the magnitude of the shunt. Anomalous pulmonary venous connection of the right upper pulmonary vein to the SVC is one of the most common and can be associated with superior SV defects ( Fig. 12.7 ,  ,,, ). Anomalous pulmonary venous connection of the right lower pulmonary vein to the IVC is associated with Scimitar syndrome which includes sequestration of the right lower lobe and hypoplasia of the right pulmonary artery. Long-term sequelae and management of PAPVC are similar to those of an ASD and are dependent on the size of the left-to-right shunt and is similar to that of an ASD. It is unusual for a single anomalous pulmonary venous connection draining one pulmonary lobe to result in a sufficient volume load to justify surgical repair. After surgical repair, TAPVC or PAPVC are at risk primarily for stenosis or thrombosis of the surgically operated pulmonary and the systemic vein specifically in PAPVC. Pulmonary vein stenosis is difficult to treat and can result in pulmonary hypertension.
,,, ). Anomalous pulmonary venous connection of the right lower pulmonary vein to the IVC is associated with Scimitar syndrome which includes sequestration of the right lower lobe and hypoplasia of the right pulmonary artery. Long-term sequelae and management of PAPVC are similar to those of an ASD and are dependent on the size of the left-to-right shunt and is similar to that of an ASD. It is unusual for a single anomalous pulmonary venous connection draining one pulmonary lobe to result in a sufficient volume load to justify surgical repair. After surgical repair, TAPVC or PAPVC are at risk primarily for stenosis or thrombosis of the surgically operated pulmonary and the systemic vein specifically in PAPVC. Pulmonary vein stenosis is difficult to treat and can result in pulmonary hypertension.
Diagnostic Strategies and Therapeutic Implications
Echocardiography remains the primary imaging modality for the diagnosis, surveillance management, and risk stratification of shunt lesions. The size, location, and physiologic manifestations of the shunt can be seen on TTE. TEE can be used for the diagnosis of intracardiac shunts in adults with suboptimal acoustic windows to guide transcatheter closure in children and adults and to assess for the presence of residual shunts after surgical closure in the perioperative period. CMR is often used in the diagnosis and surveillance of unrepaired or residual shunts in adults with suboptimal acoustic windows ( Fig. 12.16 ,  ). It remains the gold standard for the quantification of right ventricular size and the noninvasive evaluation of Qp/Qs and thus helps in determining the hemodynamic significance of the shunts. It does require sedation in young children. Cardiac CT is often used in adults with unrepaired or residual shunts to evaluate for associated anomalous pulmonary venous connections in sinus venosus defect, for the preplanning of transcatheter procedures, in detecting extracardiac shunts such as PDA when echocardiography is limited and to evaluate the physiologic manifestations of the shunt when cardiac MR is contraindicated such as in patients with pacemakers/defibrillators and other non-MRI compatible implants/foreign bodies. Cardiac catheterization is used for the quantification of pulmonary vascular resistance and transcatheter closure of shunts.
). It remains the gold standard for the quantification of right ventricular size and the noninvasive evaluation of Qp/Qs and thus helps in determining the hemodynamic significance of the shunts. It does require sedation in young children. Cardiac CT is often used in adults with unrepaired or residual shunts to evaluate for associated anomalous pulmonary venous connections in sinus venosus defect, for the preplanning of transcatheter procedures, in detecting extracardiac shunts such as PDA when echocardiography is limited and to evaluate the physiologic manifestations of the shunt when cardiac MR is contraindicated such as in patients with pacemakers/defibrillators and other non-MRI compatible implants/foreign bodies. Cardiac catheterization is used for the quantification of pulmonary vascular resistance and transcatheter closure of shunts.

Adults with surgically repaired TAPVC or PAPVC undergo surveillance for the development of pulmonary vein stenosis. Echocardiography is the most common screening modality for pulmonary vein stenosis. Color and spectral Doppler show flow acceleration across the pulmonary vein and a monophasic forward flow pattern with increased velocity. Depending on the number of pulmonary veins stenosed right ventricular hypertrophy and right ventricular dysfunction as well as elevated TR velocity can be seen suggesting pulmonary hypertension. As stenosis progresses echo may not be able to reliably detect the severity of the stenosis since flow redistributes from lung segments with obstructed pulmonary veins to unobstructed segments. Consequently, decreased pulmonary venous flow may result in a decrease in echocardiographically derived pulmonary vein gradients despite the progression of stenosis within the involved pulmonary vein. Cross-sectional imaging with computed tomography angiography (CTA) or CMR is ideal to visualize the pulmonary veins. Differential pulmonary blood flow (PBF) to the lungs and the flow in the individual pulmonary veins can be quantified by CMR. CT allows evaluation of the distal pulmonary vasculature and the lungs. Lung perfusion scans are another way to quantify perfusion to various lung segments and can provide complementary information, similar to flow measurements done with CMR. Cardiac catheterization offers visualization of the pulmonary veins throughout the lung and direct measurement of pulmonary pressures and vascular resistance. Importantly, catheterization offers potential for therapeutic interventions with balloon dilations and stent placement.
In adults with unrepaired anomalous pulmonary venous connections TTE or TEE may identify the anomalous pulmonary veins and depending on the number of pulmonary veins involved may show right atrial and right ventricular dilation. In patients who have developed pulmonary hypertension because of an unprotected pulmonary vascular bed, right ventricular hypertrophy and dysfunction can be seen on echo as well as an elevated tricuspid regurgitant jet velocity. CMR or CTA are ideal for delineating pulmonary venous connections. CMR may also quantify the degree of shunting. In patients at risk or suspected to have developed pulmonary hypertension especially if being considered for surgical correction invasive hemodynamic assessment by catheterization can be useful for direct measurement of pressures, quantification of shunt magnitude, and measurement of pulmonary arterial resistance and responsiveness to pulmonary vasodilator therapy.
Obstructive Left-Sided Heart Lesions
Clinical Overview
Congenital left-sided obstructive heart disease comprises a range of stenotic lesions that can occur in isolation or as a series of multilevel obstructions. Shone’s syndrome for example is a constellation of left-sided obstructive lesions including supravalvular mitral ring, parachute mitral valve, subaortic stenosis, coarctation, and bicuspid aortic valve. Obstruction to left ventricular inflow leads to higher atrial filling pressures, pulmonary edema, and eventually to pulmonary venous hypertension. Obstruction to left ventricular outflow causes a pressure load on the left ventricle, leading to compensatory adverse remodeling and hypertrophy diastolic and systolic dysfunction. In rare cases, adult patients may present with angina due to coronary insufficiency from increased left ventricular myocardial oxygen requirements in the hypertrophied ventricle. Severe left ventricular obstructive heart lesions are often diagnosed and repaired in childhood. Adults often present with either residual or recurrent disease after surgical repair or in mild forms of native left-sided obstruction.
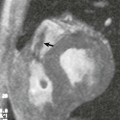
Obstruction to left ventricular inflow are rare lesions (0.1%–0.4% of all congenital heart disease) that can result in obstruction at the pulmonary venous level or at the level of the mitral valve apparatus. Cor triatriatum sinister ( Fig. 12.17 ) is an obstructive membrane that results in pulmonary venous obstruction. It divides the left atrium into a proximal chamber that receives the pulmonary veins and a distal chamber that communicates with the mitral valve and the left atrial appendage. It is often associated with ASDs and anomalous pulmonary venous connection. The obstruction is relieved by surgical resection of the membrane. Congenital stenosis of the mitral valve often involves one or more components of the mitral valve apparatus. In its classic form, the chordae tendinea are short, and thickened, the papillary muscles are closely spaced, and the leaflets are thickened and dysplastic. In an arcade mitral valve, there is almost complete obliteration of the interchordal space because of thickened and short chordae. In a parachute mitral valve there is a single papillary muscle that receives all chordal attachments ( Fig. 12.18 ,  ,, ). The other papillary muscle is either absent or underdeveloped. A double orifice mitral valve ( Fig. 12.19 ,
,, ). The other papillary muscle is either absent or underdeveloped. A double orifice mitral valve ( Fig. 12.19 ,  ) can also result in mitral stenosis resulting from abnormal tensor apparatus. Indications for intervention in mitral stenosis and left ventricular inflow lesions are described in the valvular heart guidelines. It is important to elucidate the mechanism of mitral valve stenosis and to differentiate mitral valve stenosis from supravalvar mitral ring. The latter is a fibrous membrane that is present on the left atrial side of the mitral valve and is frequently adherent to the mitral valve ( Fig. 12.20 ,
) can also result in mitral stenosis resulting from abnormal tensor apparatus. Indications for intervention in mitral stenosis and left ventricular inflow lesions are described in the valvular heart guidelines. It is important to elucidate the mechanism of mitral valve stenosis and to differentiate mitral valve stenosis from supravalvar mitral ring. The latter is a fibrous membrane that is present on the left atrial side of the mitral valve and is frequently adherent to the mitral valve ( Fig. 12.20 ,  ). Yet management between the two differs. In supravalvar mitral ring treatment involves surgical resection of the ring.
). Yet management between the two differs. In supravalvar mitral ring treatment involves surgical resection of the ring.




Obstruction to left ventricular outflow is a rare lesion (<1 out of 1000 live births) that includes subaortic stenosis, congenital aortic valve stenosis, supravalvar aortic stenosis, and coarctation. Subaortic stenosis can result from either a discrete membrane or fibrous ridge below the aortic valve or long segment narrowing of the left ventricular outflow tract or as posterior malalignment of the conal septum ( Fig. 12.21 ,  ,, ). The membranous or fibrous ridge can be surgically resected albeit with a high incidence of recurrence especially when adherent to the aortic valve leaflets. The tubular hypoplasia form is associated with a posterior malalignment VSD, aortic arch interruption type B, and coarctation. Congenital aortic valve stenosis results from a combination of commissural underdevelopment, myxomatous thickening of the valve leaflets, and or annular hypoplasia. The valve may be unicuspid or bicuspid. The management of congenital aortic valve stenosis beyond childhood in adolescents and adults is generally as directed by the 2014 valvular heart disease guidelines. Balloon aortic valvuloplasty is usually the preferred initial intervention for critical or severe aortic stenosis in infants and children. The most common form of supravalvar aortic stenosis is a discrete narrowing at the sinotubular junction ( Fig. 12.22 ,
,, ). The membranous or fibrous ridge can be surgically resected albeit with a high incidence of recurrence especially when adherent to the aortic valve leaflets. The tubular hypoplasia form is associated with a posterior malalignment VSD, aortic arch interruption type B, and coarctation. Congenital aortic valve stenosis results from a combination of commissural underdevelopment, myxomatous thickening of the valve leaflets, and or annular hypoplasia. The valve may be unicuspid or bicuspid. The management of congenital aortic valve stenosis beyond childhood in adolescents and adults is generally as directed by the 2014 valvular heart disease guidelines. Balloon aortic valvuloplasty is usually the preferred initial intervention for critical or severe aortic stenosis in infants and children. The most common form of supravalvar aortic stenosis is a discrete narrowing at the sinotubular junction ( Fig. 12.22 ,  ). It occurs frequently in patients with Williams syndrome caused by deletion in chromosome 7 resulting in defects in the elastin gene. Williams syndrome can also be associated with coronary artery stenosis, hypoplasia of the ascending aorta aortic arch and head and neck vessels, and abdominal coarctation.
). It occurs frequently in patients with Williams syndrome caused by deletion in chromosome 7 resulting in defects in the elastin gene. Williams syndrome can also be associated with coronary artery stenosis, hypoplasia of the ascending aorta aortic arch and head and neck vessels, and abdominal coarctation.


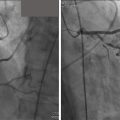
Coarctation of the Aorta and Interrupted Aortic Arch is the seventh most common congenital heart disease with one-third of patients presenting beyond infancy. It is more common in men and occurs in 12% of women with Turner syndrome. More than 50% of patients with coarctation have other associated congenital heart lesions such as bicuspid aortic valve, VSDs, other associated left-sided heart lesions, and transposition of the great arteries. In neonates, the distal transverse arch is elongated and hypoplastic resulting in a long segment coarctation yet this is less common in children and adults where the coarctation is usually discrete. In adults with unrepaired coarctation systemic collaterals develop from increased flow through the internal mammary, intercostals, and scapular arteries resulting in rib notching. Involvement of other segments of the aorta such as the abdominal aorta ( Fig. 12.23 ,  ) is rarely seen and should raise the concern for Williams syndrome or Takayasu arteritis. Interruption of the aortic arch is a rare form of ductal-dependent arch obstruction where there is a discontinuity between adjacent segments of the aortic arch. The distal segment is supplied by the PDA and the proximal segment is supplied by the ascending aorta. The interruption could be distal to the left subclavian artery (type A 30%–37%), most commonly between the left common carotid artery and the left subclavian artery (type B 62%–70%) and rarely between the carotid arteries (type C < 1%). Interruption of the aortic arch is commonly seen in patients with 22q11 microdeletion and velocardiofacial syndrome. The majority of type B interruptions are associated with a posterior malalignment ventricular septal defect (>95%). Critical or severe coarctation is repaired in the neonatal period. Adults with native coarctation present with systemic hypertension and upper to lower cuff blood pressure differential (>20 mm Hg with preserved left ventricular systolic function or >10 mm Hg with decreased left ventricular systolic function), absent or diminished femoral pulses with radiofemoral delay, and a systolic murmur in the interscapular region. Surgical repair is favored in the neonatal period by either resection of the coarctation and end-to-end anastomosis, or by reversed subclavian flap aortoplasty. Less commonly used surgical techniques include subclavian flap aortoplasty, patch plasty of the coarctation site, or placement of a bypass graft. Primary surgical anastomosis between the interrupted segments is favored in aortic arch interruption. Autologous pericardium or a prosthetic graft is limited to cases where the gap between the interrupted aortic segments cannot be fully closed. Recurrent coarctation occurs in 10% of surgically repaired coarctation. Percutaneous balloon angioplasty and endovascular stent implantation are used for the treatment of recurrent coarctation and native coarctation in older children and adults. Patients with coarctation require lifelong follow-up monitoring for systemic hypertension seen in native and repaired coarctation, recurrent coarctation, aneurysms, pseudoaneurysms, and dissection. A high incidence of aneurysms at the repair site is seen in coarctation repaired with a bypass graft. Adolescents and adults with native or repaired coarctation are at risk for intracranial aneurysms.
) is rarely seen and should raise the concern for Williams syndrome or Takayasu arteritis. Interruption of the aortic arch is a rare form of ductal-dependent arch obstruction where there is a discontinuity between adjacent segments of the aortic arch. The distal segment is supplied by the PDA and the proximal segment is supplied by the ascending aorta. The interruption could be distal to the left subclavian artery (type A 30%–37%), most commonly between the left common carotid artery and the left subclavian artery (type B 62%–70%) and rarely between the carotid arteries (type C < 1%). Interruption of the aortic arch is commonly seen in patients with 22q11 microdeletion and velocardiofacial syndrome. The majority of type B interruptions are associated with a posterior malalignment ventricular septal defect (>95%). Critical or severe coarctation is repaired in the neonatal period. Adults with native coarctation present with systemic hypertension and upper to lower cuff blood pressure differential (>20 mm Hg with preserved left ventricular systolic function or >10 mm Hg with decreased left ventricular systolic function), absent or diminished femoral pulses with radiofemoral delay, and a systolic murmur in the interscapular region. Surgical repair is favored in the neonatal period by either resection of the coarctation and end-to-end anastomosis, or by reversed subclavian flap aortoplasty. Less commonly used surgical techniques include subclavian flap aortoplasty, patch plasty of the coarctation site, or placement of a bypass graft. Primary surgical anastomosis between the interrupted segments is favored in aortic arch interruption. Autologous pericardium or a prosthetic graft is limited to cases where the gap between the interrupted aortic segments cannot be fully closed. Recurrent coarctation occurs in 10% of surgically repaired coarctation. Percutaneous balloon angioplasty and endovascular stent implantation are used for the treatment of recurrent coarctation and native coarctation in older children and adults. Patients with coarctation require lifelong follow-up monitoring for systemic hypertension seen in native and repaired coarctation, recurrent coarctation, aneurysms, pseudoaneurysms, and dissection. A high incidence of aneurysms at the repair site is seen in coarctation repaired with a bypass graft. Adolescents and adults with native or repaired coarctation are at risk for intracranial aneurysms.