Congenital ear or temporal bone malformations are a diagnostic challenge to radiologists and surgeons alike. Newer imaging techniques can detect subtle changes in middle ear and cochlear anatomy. This information is invaluable with increasing use of hearing restoration surgeries and/or cochlear implants in such patients. This article discusses the embryogenesis, classification system, and salient imaging findings of congenital outer, middle ear, and inner ear anomalies in children. Both high-resolution computerized tomography and magnetic resonance imaging scans of the temporal bones are described.
Cochlear implants have revolutionized the thinking and approach to congenitally deaf children. More and more otologists are now performing surgeries on the malformed ear, especially for malformed cochleae. Faster, safer, and better imaging techniques have greatly contributed to understanding of the ear anatomy. Although bony abnormalities of the labyrinth account for only 20% of all cases of congenital deafness, such information is of practical importance to a dedicated cochlear implant team that is likely to encounter these uncommon cases. Recognition of these malformations forewarns a surgeon to the possibility of potential complications and guides parental expectations during preoperative counseling. Similarly, imaging provides vital information about external and middle ear malformations to the surgeons. This article provides a common portal for both neurotologists and neuroradiologists to understand the wide spectrum of ear malformations, gain insight into their embryogenesis, formulate correct candidacy decisions, and also become equipped with the clinical knowledge of what to expect when encountering a malformed ear.
Embryology of ear structures
The temporal bone has 2 separate precursors. The pars branchialis forms from the first and second branchial arches, the first branchial cleft, and the adjacent mesenchyme, and subsequently forms the external ear and middle ear structures. The pars otica develops from the otic vesicle and gives rise to the inner ear structures. The development of the inner ear is therefore largely independent of the development of the external and middle ear.
The pinna or the auricle develops from the ectoderm of the first (mandibular) and second (hyoid) arches. Development begins around the 40th to 45th day and is completed by fourth month of fetal life. The external auditory canal (EAC) arises as a shallow pit in the first branchial cleft. Development begins around the sixth fetal week and is completed by the seventh month. The invaginations of the first branchial cleft and the first branchial pouch with the associated mesenchyme result in the formation of the primitive tympanic membrane. The tubotympanic recess develops from the first branchial pouch between weeks 4 and 30 and gives rise to the eustachian tube, tympanic cavity, and mastoid antrum ( Fig. 1 ). The middle ear ossicles, except stapes footplate, arise from the mesenchyme of the first and second branchial arches. The stapes footplate and annular ligament develop from the otic capsule.
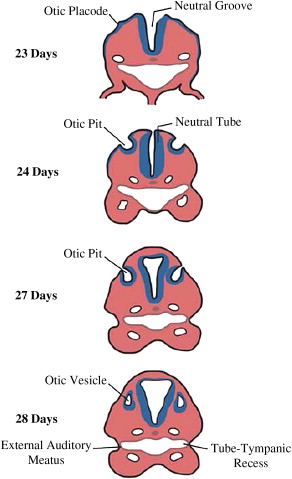
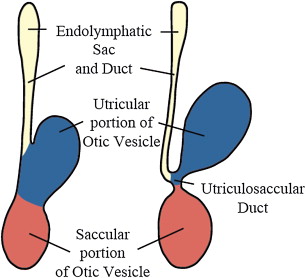
The inner ear develops between the fourth and eighth week of gestation and ossifies as the temporal bone between the 16th and 24th weeks. The membranous cochlea begins to develop at 22 days as the otic placode (see Fig. 1 ); this invaginates into the surrounding mesenchyme and cochlear development (including bony and membranous coils) and is completed by the eighth week. The saccule and the utricle are completely formed by the 11th week of gestation ( Fig. 2 ). The semicircular canals (SCCs) develop from the vestibular anlage between the sixth and eighth weeks and are completely formed by the 22nd week of gestation. The superior SCC develops first, followed by the posterior and the lateral SCC. Thus, developmental abnormalities before the eighth week may lead to various cochlear abnormalities depending on the stage of arrest of embryogenesis. Problems occurring between weeks 8 and 11 of gestation can affect the saccule, utricle, and/or the SCC with normal cochlea. Because the lateral SCC is the last to develop, it is most commonly affected.
Incidence
Congenital malformations or dysplasias are distinct from anatomic variants because malformations have both abnormal anatomy and altered function. Malformations of the external ear are referred to as congenital aural dysplasias. Malformations of the auricle alone are called microtias. Congenital aural dysplasias have been reported to occur in 1:3000 to 1:10,000 births. Most cases are unilateral, without other associated systemic abnormalities and without a known cause. In unilateral cases, the right side is most commonly affected (58%–61%). Microtia has been reported to occur predominantly in boys, with a male/female ratio of 2:1. Congenital aural dysplasias may occur in association with genetic disorders, chromosomal anomalies, and intrauterine infections. Common syndromes associated include Treacher Collins, Goldenhar, and Klippel-Fiel syndromes. Overall, combined congenital malformation of the external and middle ear occur more commonly than isolated middle ear anomalies. Associated inner ear malformations are less common (10%) because of the separate embryogenesis of the labyrinth.
A study by Westerhof and colleagues revealed that most patients with inner ear anomalies had multiple abnormalities. The most common malformations of the inner ear include anomalies of the SCC (88%) followed by malformations of the vestibule (62%). Enlarged vestibular aqueduct is the most common isolated malformation. Among cochlear anomalies, the classic Mondini deformity accounts for nearly 55%, whereas common cavity malformations are seen in 25% of patients. Syndromes such as CHARGE (coloboma, heart defects, choanal atresia, retarded growth and development, genital malformations and ear anomalies), Pendred, branchio-oto-renal (BOR), Phelps X-linked mixed deafness, Waardenburgh, and others are associated with inner ear malformations but are less common than nonsyndromic congenital bony abnormalities. Table 1 summarizes the cochlear malformations, the gestational age of developmental arrest, and their frequency as reported in literature.
| Cochlear Deformities | Gestational Age at Arrest (wk) | Frequency |
|---|---|---|
| Michel deformity | Third | Very rare |
| Common cavity | Between fourth and fifth | 25% |
| Cochlear aplasia | Fifth | Uncommon |
| Cochlear hypoplasia | Sixth | 15% |
| IP I (pseudo-Mondini) | Between sixth and seventh | Uncommon |
| IP II (classic Mondini) | Seventh | 55% |
Incidence
Congenital malformations or dysplasias are distinct from anatomic variants because malformations have both abnormal anatomy and altered function. Malformations of the external ear are referred to as congenital aural dysplasias. Malformations of the auricle alone are called microtias. Congenital aural dysplasias have been reported to occur in 1:3000 to 1:10,000 births. Most cases are unilateral, without other associated systemic abnormalities and without a known cause. In unilateral cases, the right side is most commonly affected (58%–61%). Microtia has been reported to occur predominantly in boys, with a male/female ratio of 2:1. Congenital aural dysplasias may occur in association with genetic disorders, chromosomal anomalies, and intrauterine infections. Common syndromes associated include Treacher Collins, Goldenhar, and Klippel-Fiel syndromes. Overall, combined congenital malformation of the external and middle ear occur more commonly than isolated middle ear anomalies. Associated inner ear malformations are less common (10%) because of the separate embryogenesis of the labyrinth.
A study by Westerhof and colleagues revealed that most patients with inner ear anomalies had multiple abnormalities. The most common malformations of the inner ear include anomalies of the SCC (88%) followed by malformations of the vestibule (62%). Enlarged vestibular aqueduct is the most common isolated malformation. Among cochlear anomalies, the classic Mondini deformity accounts for nearly 55%, whereas common cavity malformations are seen in 25% of patients. Syndromes such as CHARGE (coloboma, heart defects, choanal atresia, retarded growth and development, genital malformations and ear anomalies), Pendred, branchio-oto-renal (BOR), Phelps X-linked mixed deafness, Waardenburgh, and others are associated with inner ear malformations but are less common than nonsyndromic congenital bony abnormalities. Table 1 summarizes the cochlear malformations, the gestational age of developmental arrest, and their frequency as reported in literature.
| Cochlear Deformities | Gestational Age at Arrest (wk) | Frequency |
|---|---|---|
| Michel deformity | Third | Very rare |
| Common cavity | Between fourth and fifth | 25% |
| Cochlear aplasia | Fifth | Uncommon |
| Cochlear hypoplasia | Sixth | 15% |
| IP I (pseudo-Mondini) | Between sixth and seventh | Uncommon |
| IP II (classic Mondini) | Seventh | 55% |
Imaging techniques for temporal bone malformations
Computed tomography (CT) is the imaging method of choice to detect anomalies in congenital aural dysplasia. For best resolution, the slice thickness should be submillimeter with isovoxel imaging, bone algorithm, and a small field of view. Multidetector CT is not only fast but also allows excellent coronal reconstruction, obviating the need to scan in a second plane. Imaging for congenital aural dysplasia is not only important to confirm the presence or absence of malformations but is also required to grade the degree and severity of the malformation. This information is invaluable to a surgeon and serves as a basis for treatment decisions and outcomes. A surgical rating scale has been developed by Jahrsdoerfer and colleagues from correlating clinical findings and CT images. The grading system is based on a possible score of 10. The stapes is assigned the highest rating (2 points) and the rest of the structures are awarded 1 point each. The higher the rating, the better are the chances of successful hearing restoration after surgery. Patients who have a score of 5/10 or less have been shown to not benefit from surgery. For children with congenital sensorineural deafness, both axial and coronal CT views and axial MR imaging views are the usual standard. CT enables simultaneous visualization of associated middle and external ear abnormalities. It provides knowledge of the thickness of the parietal bone, degree of pneumatization of mastoid air cells, and presence of retrocochlear and infracochlear air cells that may be mistaken for the round window niche. MR imaging is performed to assess the membranous labyrinth, internal auditory canal (IAC), and cerebellopontine angle. T2-weighted sequence or fluid-attenuated inversion recovery (FLAIR) for the brain to assess the auditory cortex is also required. Several thin gradient echo images are required, which can be achieved by T2 heavily weighted sequences like three-dimensional (3D) Fourier transform constructive interference in steady state (3DFT-CISS), true fast imaging with steady precession (FISP), or 3D driven enhancement (3D-T2 DRIVE). Gadolinium is required to assess enhancement of the membranous labyrinth in case of postmeningitic or autoimmune labyrinthitis. Specific advantages of MR imaging include (1) detection of an existing cochlear nerve; (2) demarcation of cochlear and labyrinthine structures, which is essential to ensure correct insertion of the electrode array during surgery; (3) early detection of postmeningitis fibro-osseous change in the cochlea; and (4) evaluation of associated abnormalities within the brainstem and central auditory pathways. Plain radiographs do not have any role in the assessment of potential cochlear malformations, but are important after surgery in assessing the position and depth of electrode insertion. A modified Stenver view shows the typical coiled appearance of a fully inserted electrode array. An intraoperative per orbital view using an image intensifier is required when there is doubt regarding correct placement of electrodes.
Anomalies of the external ear
Auricle
Auricular malformations can affect the size, shape, position, and orientation of the pinna. Complete absence of the pinna (anotia) may occur. Various classification schemes have been proposed to grade the severity of deformity; however, Weerda’s classification is most widely used. This classification includes 3 grades of microtia with worsening severity. The important aspects of this classification have been summarized in Table 2 .
| Grade of Dysplasia | Characteristics |
|---|---|
| Grade I (slight malformation) | Most structures of the normal pinna are recognizable. Surgery usually does not require use of additional skin or cartilage |
| Grade II (moderate malformation) | Some structures of a normal pinna are recognizable. Surgery involves use of additional skin or cartilage |
| Grade III (severe malformation) | None of the normal structures of the pinna are recognizable. Includes anotia. Total reconstruction is necessary, which requires much of the adjacent skin and cartilage |
EAC
Failure of recanalization of tissue of the first branchial cleft can lead to EAC malformations. The absence of a meatal opening under the tragus signifies complete aural atresia ( Fig. 3 ). Incomplete atresia or stenosis should be suspected when the pinna is abnormal and the EAC diameter is less than 4 mm or the tympanic membrane cannot be visualized. The stenosis can be fibrous, bony, or both. In the fibrous type, there is a soft tissue plug at the position of the tympanic membrane, whereas the bony stenosis is characterized by the presence of a bony plate at the level of the tympanic membrane ( Fig. 4 A). Schuknecht has classified EAC atresia into 4 types (A–D) and this classification is useful to both surgeons and the radiologists. Weerda has classified EAC malformations into 3 types (A–C) and these are described in Table 3 . With abnormal EAC formation, the structures around the ear are also displaced from their normal position. The condylar fossa is higher than usual and posteriorly displaced, whereas the mastoid process is pushed anteriorly. The jugular bulb may be high and the tegmen tympani low. The facial nerve has an anomalous course in patients with congenital aural dysplasias. The tympanic and mastoid segments are most commonly affected. The tympanic segment is typically displaced inferiorly at the level of the round window, and may be dehiscent or displaced medially to overlie the oval window. The mastoid segment is shallow and displaced anterolaterally, exiting the temporal bone at the level of the round window (see Fig. 4 B).
| Types of EAC Stenosis | Characteristics |
|---|---|
| Type A | Marked narrowing of the EAC with an intact skin layer |
| Type B | Partial development of the EAC with an atresia plate medially |
| Type C | Complete bony EAC stenosis |
First Branchial Cleft Anomalies
These occur as a result of abnormal ectodermal closure of the cleft. They typically occur along the line from the floor of the EAC to the submental area. These anomalies may present as preauricular cyst, sinus, asymptomatic or infected parotid mass, or recurrent external otitis. Please refer to the article by Ibrahim and colleagues elsewhere in this issue for further details on this entity.
Congenital Cholesteatoma
Congenital cholesteatoma occurs more commonly in the middle ear and is also discussed later in this article. Children with atresia of the EAC may develop cholesteatomas (primary or secondary) at the site of the stenosis or deep to the atretic plate.
Anomalies of the middle ear
It is generally known that the better developed the auricle and external ear the better developed the middle ear. A study by Ishimoto and colleagues analyzing CT grading system for middle ear abnormalities versus severity of microtia suggested an inverse relationship between the two (ie, the better the CT score, the less the severity of microtia). Middle ear malformations can affect the normal development of the tympanic cavity, mastoid pneumatization, and ossicles. There are various classifications proposed, so it is important that the radiologist and the otologist use the same or similar classification to evaluate middle ear anatomy.
Anomalies of Ossicles and Related Structures
Malleus
These include aplasia, fixation of the incudomalleolar joint, bony fusion of the head of the malleus to the long process of the incus and stapes head (triple bony union), and deformed head. A rare anomaly of the malleus is congenital fixation of the malleus head to the lateral epitympanic wall called the malleus bar ( Fig. 5 ).
Incus
These include aplasia, deformity of the long process, fusion of short process of incus to lateral SCC, and fibrous union of the incudostapedial joint or absence of the joint.
Stapes
These include absence of the stapes suprastructure, aplasia, deformed head, monopod stapes ( Fig. 6 ), fixation of head to promontory, and footplate fixation.
Related posts:
 Demystifying Congenital Malformations for Imaging Specialists
Demystifying Congenital Malformations for Imaging Specialists
 Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
 Neurosurgical Management of Congenital Malformations of the Brain
Neurosurgical Management of Congenital Malformations of the Brain
 Imaging of Congenital Spine and Spinal Cord Malformations
Imaging of Congenital Spine and Spinal Cord Malformations
 Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
 Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


