7 Constitutional Disorders of Bone Growth The constitutional disorders of bone growth encompass a vast number of congenital conditions associated with disturbed bone growth and development in all or parts of the skeleton. This category of disorders includes more than 100 well-defined entities and more than 1000 not-yet-categorized symptom complexes. The original summary and categorization of constitutional disorders of bone were described in the Paris Nomenclature of 1986, and the latest revision was reported in 1992. According to this nomenclature, the category of constitutional disorders of bone growth includes skeletal or osteochondrodysplastic dysplasias, primary metabolic disorders, chromosomal aberrations, dysostoses, and idiopathic osteolyses. Dysostosis is an abnormal development of individual bones, isolated or combined. The term ‘osteodystrophy’ has been abandoned. This chapter describes the most frequent conditions selected from the vast number of congenital skeletal conditions, and outlines the most characteristic features of these diseases for better detection and appropriate categorization. The selection criteria in Table 7.1 follow the International Nomenclature and Caffey’s designation. Overlapping of the proposed classifying features is unavoidable. The literature should be consulted for detailed information related to the various entities. Synonym: Osteochondrodysplasias The important clinical manifestation of osteochondrodysplasias is inferior bone growth. The height reached in adulthood generally is below 140 cm (approximately 55 inches). The disturbance in bone growth is disproportional and, depending on the relative involvement of the spine or trunk, the trunk or the limbs are shortened. Disproportionate limb growth can lead to rhizomelic (proximal), mesomelic (medial) or acromelic (distal) types of micromelia. Kyphoscoliosis, deformities of the extremities, and misalignment of the joints are prominent features. Diagnosing skeletal dysplasias is often difficult and relies on clinical, genetic, histologic, and histochemical methods. Radiographs are very important and often essential for the diagnosis. Discriminating radiographic features are deviation in size, shape, and bone density of particular skeletal segments. A singular anomaly generally excludes a skeletal dysplasia, but two or more identified deviations might suffice for diagnostic assignment to a skeletal dysplasia. The prerequisite is the systematic analysis of the radiographic features to address the following questions: The radiographic examination of patients with suspected skeletal dysplasia consists of three or four views of the most frequently involved skeletal sites. Supplementary views should be added if needed. Survey radiographic views:
 Skeletal dysplasia (synonym: osteochon-drodysplasia) is a generalized abnormality of cartilage and bone development.
Skeletal dysplasia (synonym: osteochon-drodysplasia) is a generalized abnormality of cartilage and bone development.

Skeletal Dysplasias
I. Skeletal dysplasias |
A. Identifiable at birth |
1. Thanatophoric dysplasia |
2. Achondroplasia |
3. Chondrodysplasia punctata |
3.1 Rhizomelicform |
3.2 Conradi-Hünermann form |
4. Asphyxiating thoracic dysplasia |
5. Dyschondrosteosis (Léri-Weill disease) |
6. Cleidocranial dysplasia |
B. Identifiable later in life |
Predominantly epiphyseal dysplasias: |
1. Multiple epiphyseal dysplasia |
Predominantly metaphyseal dysplasias |
2. Hypochondroplasia |
3. Metaphyseal chondrodysplasias: |
3.1 Schmid type |
3.2 McKusick type |
3.3 Schwachman type |
Predominantly spondyloepiphyseal or metaphyseal dysplasias: |
4. Spondyloepimetaphyseal dysplasia congenita |
5. Spondyloepimetaphyseal dysplasia tarda |
II. Dysostosis multiplex group |
1. Mucopolysaccharidoses |
2. Mucolipidoses, gangliosidoses |
III. Dysostoses |
1. With predominant craniofacial involvement |
2. With predominant axial involvement |
3. With predominant limb involvement |
IV. Idiopathic osteolyses (very rare) |
C. Skeletal dysplasia with disorganized development of cartilage and fibrous components |
1. Multiple cartilaginous exostoses (Diaphyseal aclasis) |
2. Enchondromatosis (Oilier) |
3. Polyostotic fibrous dysplasia |
4. Congenital generalized fibromatosis |
D. Abnormal density of osseous structures |
Decreased bone density: |
1. Osteogenesis imperfecta |
2. Hypophosphatasia |
3. Hereditary rickets |
3.1 Phosphate diabetes |
3.2 Pseudodeficiency rickets |
Increased bone density: |
4. Osteopetrosis |
5. Pyknodysostosis |
6. Osteopoikilosis |
7. Osteopathia striata |
8. Melorheostosis |
Early Onset Types
Many skeletal dysplasias can be diagnosed in utero or in the neonatal period. These are characterized by a disproportion between the head, trunk, and limbs. Identifying fetal disproportions by obstetrical sonography can suggest a skeletal dysplasia. Stillbirths or death in the neonatal period due to associated anomalies with impaired vital functions is common. Postnatally, the radiographic evaluation is crucial for establishing the diagnosis. A definitive diagnosis is mandatory for the genetic counseling of parents who wish to have more children.
Thanatophoric Dysplasia
 Mode of inheritance: Autosomal dominant with spontaneous mutation.
Mode of inheritance: Autosomal dominant with spontaneous mutation.
Prevalence: Occurring in 1:26000 to 1:58000 births, this is the most common lethal skeletal dysplasia. Its name means ‘death bearing,’ referring to the incompatibility with life due to the severe narrowing of the thorax.
Manifestation: Growth disturbance is disproportionate, as the trunk is of normal length and the extremities shortened. The thorax is extremely narrow, and the head is large with a depressed nasal root. Affected neonates that are not stillborn die within a few days after birth.
 Attention should be directed to the characteristic changes of the bony thorax, axial skeleton, and extremities (Figs. 7.1, 7.2):
Attention should be directed to the characteristic changes of the bony thorax, axial skeleton, and extremities (Figs. 7.1, 7.2):
- Extremely small bony thorax with very short ribs,
- Markedly flattened vertebral bodies with ossification defects in the end plates. Ratio of disk spaces to vertebral bodies is 3:1 (normally 1:1),
- Height of the ilium decreased, giving squared iliac wings,
- Extreme micromelia with concomitant broadening of the shaft. Widened metaphyses with flaring and cupping. So-called ‘French telephone’ deformity of the curved and shortened femur,
- Prominent cranial vault, possibly combined with a severely disturbed ossification of the sutures, producing the so-called ‘cloverleaf skull’.
 Similar but less-extensive skeletal changes are observed in achondroplasia, which has less-pronounced vertebral flattening and less shortening and bowing of the long tubular bones. Achondrogenesis has even more poorly developed and often absent ossification centers.
Similar but less-extensive skeletal changes are observed in achondroplasia, which has less-pronounced vertebral flattening and less shortening and bowing of the long tubular bones. Achondrogenesis has even more poorly developed and often absent ossification centers.
Achondroplasia
 Mode of inheritance: Autosomal dominant with a spontaneous mutation rate of 80%.
Mode of inheritance: Autosomal dominant with a spontaneous mutation rate of 80%.
Prevalence: Occurring in 1:13 500 live births, it is the most common skeletal dysplasia. The synonym ‘fetal chondrodysplasia’ has been abandoned, but the now-accepted term achondroplasia is strictly not appropriate either, since cartilage is formed.
Manifestation: Affected individuals show disproportionate, micromelic, and rhizomelic dwarfism with a final body height not exceeding 122 cm (approximately 48 inches). The head is enlarged and shows a prominent forehead and a flattened nasal bridge. Characteristic features are a thoracolumbar kyphosis and protruding buttocks due to an increased pelvic tilt. The fingers are shortened and have a three-pronged or trident appearance.
 Attention should be directed to the characteristic deformities of the pelvis and posterior vertebral arches, to the disproportionately short extremities, and to the severely disturbed ossification in the epiphyseal growth zone:
Attention should be directed to the characteristic deformities of the pelvis and posterior vertebral arches, to the disproportionately short extremities, and to the severely disturbed ossification in the epiphyseal growth zone:
- Squared pelvis (‘elephant ears’) already present in early childhood; an extremely small sacroiliac notch and an almost horizontally oriented acetabular roof (Fig. 7.3),
- Short and broad tubular bones with rhizomelic exaggeration; characteristic oval radiolucency of the upper ends of the femur in neonates; widened and medially enlarged distal femoral metaphyses (Fig. 7.4); partial metaphyseal invagination of the epiphyses, so-called ‘ball-in-socket epiphyseal-metaphyseal junction,’
- Short and flattened vertebral bodies with posterior scalloping; progressive decrease in the inter-pediculate distance of the lumbar spine from cranial to caudal (Fig. 7.5),
- Large cranial vault resting on a small cranial base, constricted foramen magnum and dilated CSF space by CT,
- ‘Overlapping fibula’ due to overgrowth of the fibula relative to the tibia.
 The common heterozygotic type has easily recognized features, whereas the less common homozygotic type resembles thanatophoric dysplasia; the changes of hypochondroplasia are variable and less severe.
The common heterozygotic type has easily recognized features, whereas the less common homozygotic type resembles thanatophoric dysplasia; the changes of hypochondroplasia are variable and less severe.
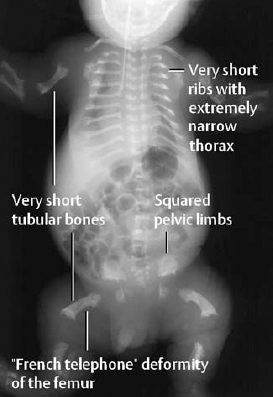
Fig. 7.1 ‘Babygram’ of thana-tophoric dysplasia.

Fig. 7.2 Lateral spine of thana-tophoric dysplasia in the same child as in Fig. 7.1.
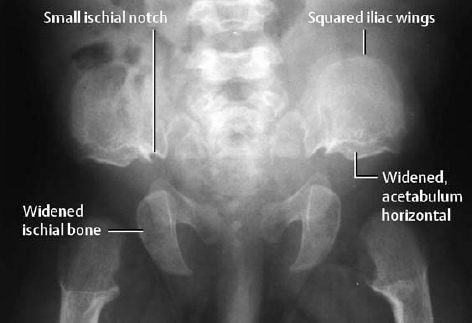
Fig. 7.3 Pelvis of a 5-month-old child with achondroplasia.
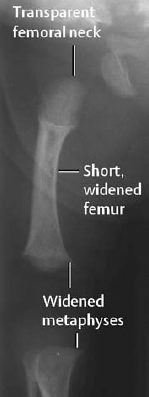
Fig. 7.4 Achondroplasia, same child as in Fig. 7.3.
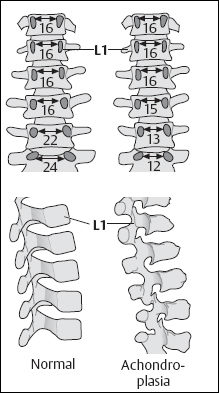
Fig. 7.5 Differences in interpediculate distances and the configuration of the vertebral bodies in a normal child and in a child with achondroplasia (after Caffey).
Chondrodysplasia Punctata
Stippled calcifications seen in the growth zones of the long tubular bones, in the axial skeleton and in the tracheal cartilage give this condition its name. Fifteen different types of dysplasia with these radiographic features have been differentiated so far. The most frequent types are the rhizomelic type and the Conradi-Hünermann type.
 Mode of inheritance: The rhizomelic type is autosomal recessive, and the Conradi-Hünermann type is X-linked dominant.
Mode of inheritance: The rhizomelic type is autosomal recessive, and the Conradi-Hünermann type is X-linked dominant.
Manifestation: Whereas the rhizomelic type has symmetric rhizomelic limb shortening, the Conradi-Hünermann type has asymmetric limb shortening. Both types are associated with craniofacial dysmorphism and cataracts.
Prognosis: The rhizomelic type follows a more severe clinical course, and death usually occurs within the first year of life due to severe infections and cerebral damage.
 Attention should be directed to the characteristic stippled calcifications in the skeletal cartilage of the newborn and, later in life, to the various manifestations of limb shortening:
Attention should be directed to the characteristic stippled calcifications in the skeletal cartilage of the newborn and, later in life, to the various manifestations of limb shortening:
- stippled calcifications in the cartilage of the extremities and, in the Conradi-Hünermann type, also in the axial skeleton, larynx, trachea, and joint capsule; disappearance of the stippled calcifications within the first year of life (Fig. 7.6),
- symmetric shortening of the long tubular bones, especially of the humeri and femora in the rhizomelic type, and asymmetric limb shortening in the Conradi-Hünermann type,
- division of the vertebral bodies in the lateral projection by a so-called ‘coronal cleft/which slowly regresses later in life.
 Severely abnormal myelinization with cortical atrophy in the rhizomelic type.
Severely abnormal myelinization with cortical atrophy in the rhizomelic type.
 This technique can directly display the relationship between the stippled calcifications and the epimetaphyseal growth zone (Fig. 7.7).
This technique can directly display the relationship between the stippled calcifications and the epimetaphyseal growth zone (Fig. 7.7).
 Other types of chondrodysplasia punctata should be included in the differential diagnosis, including fetal warfarin syndrome in infants born to mothers who took coumarin derivatives during pregnancy, fetal alcohol syndrome, and Zellweger syndrome.
Other types of chondrodysplasia punctata should be included in the differential diagnosis, including fetal warfarin syndrome in infants born to mothers who took coumarin derivatives during pregnancy, fetal alcohol syndrome, and Zellweger syndrome.
Asphyxiating Thoracic Dysplasia
Synonyms: Jeune syndrome, thoraco-pelvic-phalangeal dysplasia.
 Mode of inheritance: Autosomal recessive.
Mode of inheritance: Autosomal recessive.
Prevalence: 1:100000 live births.
This condition has a broad clinical spectrum ranging from the lethal type in neonates with a narrow thorax to the mild type without respiratory difficulties. The outstanding findings are short-limbed dwarfism and Polydactyly (in some instances). Progressive nephropathy develops later in life.
 Attention should be directed to the characteristic changes of the bony thorax and pelvis (Fig. 7.8):
Attention should be directed to the characteristic changes of the bony thorax and pelvis (Fig. 7.8):
- narrow, bell-shaped thorax with very short ribs that show parasternal flaring,
- squared pelvis with shortened sacroiliac notch and an inferior spur,
- micromelia of the long and short tubular bones with irregular metaphyses.
 In the newborn period, all skeletal dysplasias associated with a narrow thorax enter the differential diagnosis and include Ellis-van Creveld syndrome, thanatophoric dysplasia, and achondroplasia, and, in later infancy, all of the skeletal dysplasias with predominantly metaphyseal growth disturbances.
In the newborn period, all skeletal dysplasias associated with a narrow thorax enter the differential diagnosis and include Ellis-van Creveld syndrome, thanatophoric dysplasia, and achondroplasia, and, in later infancy, all of the skeletal dysplasias with predominantly metaphyseal growth disturbances.
Dyschondrosteosis
Synonym: Léri-Weill disease
 Mode of inheritance: Autosomal dominant.
Mode of inheritance: Autosomal dominant.
Manifestation: Mesomelic dwarfism with shortened forearms combined with Madelung deformity.
 Attention should be directed to the characteristic shortening and bowing of the forearm with Madelung deformity:
Attention should be directed to the characteristic shortening and bowing of the forearm with Madelung deformity:
- shortened radius (Fig. 7.9),
- ulnar and radial epiphyses are slanted toward each other with posterior dislocation of the ulna (Madelung deformity, Fig. 7.10),
- Tibial shortening may be present in some instances.
 Other mesomelic growth disturbances with shortened bones of forearm and lower leg should be taken into consideration. Turner syndrome can also show similar changes.
Other mesomelic growth disturbances with shortened bones of forearm and lower leg should be taken into consideration. Turner syndrome can also show similar changes.
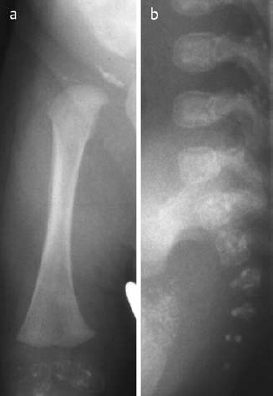
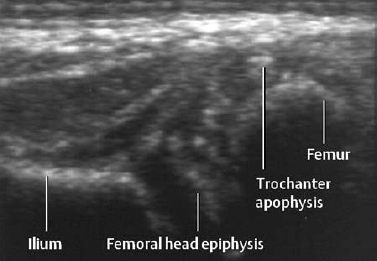
Fig. 7.7 Lateral section of sonogram in same child as in Fig. 7.6. Note the small coarse calcifications in the cartilaginous epiphysis of the femoral head and in the apophysis of the trochanter.
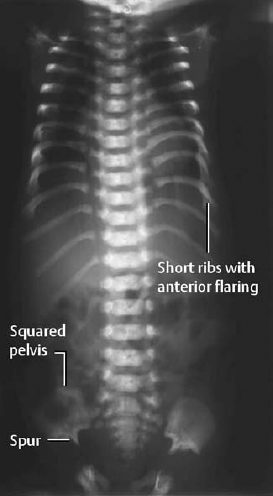
Fig. 7.8 Asphyxiating thoracic dysplasia in a newborn: AP radiograph of the trunk (babygram) shows short ribs with resultant very narrow thorax.
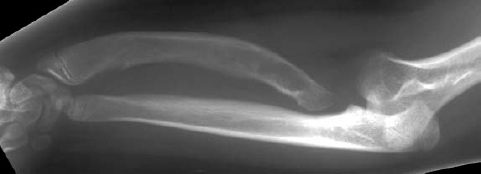
Fig. 7.9 Dyschondrosteosis in an 11-year-old girl. The forearm is shortened and the radius bowed.
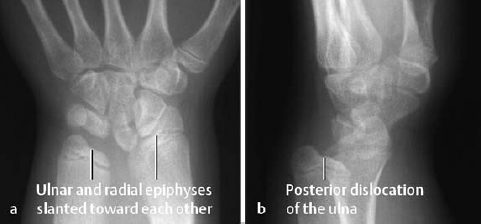
Fig. 7.10 Dyschondrosteosis in the same child as in Fig. 7.9. The distal articular surfaces of the radius and ulna are slanted toward each other. The ulna is posteriorly dislocated.
Cleidocranial Dysplasia
Synonym: Pelvico-cleidocranial dysostosis.
 Mode of inheritance: Autosomal dominant.
Mode of inheritance: Autosomal dominant.
Manifestation: The outstanding findings are the large head with an open or late-fusing anterior fontanel, disturbance of teeth development, and anterior approximation of the shoulders in cases with aplastic or hypoplastic clavicles.
 Attention should be directed to the characteristically delayed ossification of the cranial vault and to the clavicular aplasia or hypoplasia:
Attention should be directed to the characteristically delayed ossification of the cranial vault and to the clavicular aplasia or hypoplasia:
- widely patent anterior fontanelle and sutures with multiple intercalary bones (Fig. 7.11),
- aplasia or hypoplasia of both clavicles or pseudarthrosis of hypoplastic clavicles (Fig. 7.12
Related posts:

Stay updated, free articles. Join our Telegram channel



Full access? Get Clinical Tree


