Fig. 15.1
Ionotropic and metabotropic glutamate receptors (Source: http://www.ucl.ac.uk/ioo/research/salt/glutamat.htm (Prof. Tom Salt))
The iGluRs are the principal mediators of excitatory neurotransmission. These are ligand-gated cationic-selective channels which are permeable to Na+, K+, and Ca2+ ions. They are subdivided into three major classes based on structural similarities and named according to the type of synthetic agonist that activates them. These three selective agonists are: NMDA (N-methyl-d-aspartate), AMPA (α-amino-3-hydroxy-5-methyl-isoxazole-4-propionate), and kainate (Bettler and Mulle 1995; Mori and Mishina 1995). Native receptors of all of these families are likely heteromeric assemblies comprising more than one type of subunit. N-methyl-d-aspartate (NMDA) tetrameric receptors consist of subunits NR1 and NR2. There are four NR2 subunit types, NR2A, B, C, and D. AMPA receptors are composed of a four-subunit family (GluR1–4) with members that are products of separate genes and are believed to assemble as functional tetramers. Kainate receptors are composed of two related subunit families, GluR5–7 and KA-1 and 2.
Metabotropic glutamate receptors (mGluRs) are G-protein-coupled receptors (GPCR), which activate intracellular secondary messenger systems when bound by the physiological ligand glutamate. From a structural viewpoint, mGluRs belong to the seven-transmembrane protein family and have been classified into three subgroups. Group I mGluRs (mGluR1 and mGluR5) are coupled to phospholipase C and upregulate or downregulate neuronal excitability (Gereau and Conn 1995). Group II (mGluR2 and mGluR3) and group III (mGluR4 and mGluR6–8) inhibit adenylate cyclase and hence reduce synaptic transmission (Bradley et al. 2000; Ritzen et al. 2005). The mGluRs are expressed on neuronal and glial cells, with each receptor subtype exhibiting distinct spatial and temporal expression profiles in the brain (Shigemoto et al. 1997; Thomas et al. 2001), with the exception of mGluR6 existing in the retina. In neurons, group I mGluRs are mainly localized in somatodendritic domains and postsynaptically regulate neuronal excitability and synaptic transmission via several intracellular second messenger systems, whereas group II and III mGluRs are predominantly localized in axonal domains and axon terminals to presynaptically regulate neurotransmitter release (Cartmell and Schoepp 2000). MGluRs function as dimers, with two glutamate molecules being required for full receptor activation (Tsuchiya et al. 2002; Shin et al. 2008; Ono et al. 2012).
15.1.2 PET
Positron emission tomography (PET) is a noninvasive imaging technology, allowing for the quantitative imaging of physiologic and pathophysiologic processes in vivo (Talbot and Laruelle 2002; Schubiger et al. 2007; Ametamey et al. 2008). High resolution of about 1–2 mm can be achieved with small animal imaging systems and 4–6 mm for clinical PET scanners. The visualization of biological targets and systems is of special interest in this research field. Specific PET tracers for labeling such targets are potential future diagnostics of various diseases and provide elegant tools for drug development (Fowler et al. 1999), especially for the development of drugs with less side effects. Besides oncologic and cardiac applications, there is a growing demand for specific and selective radiopharmaceuticals in the field of neuroscience. PET can be used to image receptor distribution, concentration, and functions in normal and pathological states and has proven to be a powerful technique in neurological research (Passchier et al. 2002). Due to the convenient half-lives of carbon-11 and fluorine-18 and their chemical properties, most effort has been put into the development of tracers labeled with one of these isotopes. In general, carbon-11 and fluorine-18 are produced by nuclear reactions which require the bombardment of convenient stable isotopes with high energetic protons provided by a cyclotron. Carbon-11 can easily be introduced into biologically interesting compounds without changing their biochemical and pharmacological properties. The relatively short physical half-life of 11C (20 min) allows for multiple imaging studies in a short period of time, which makes it advantageous for sequential investigations with short time intervals in the same individual (animal or human), thereby allowing the subject to serve as its own control. Therefore 11C-labeled compounds can be used for instance in a receptor occupancy paradigm or in cognitive intervention studies where several PET investigations are usually needed within a few hours apart from each other. The short half-life of 11C, however, is disadvantageous for commercial production. Fluorine-18 has long physical half-life of 110 min allowing more complex synthesis, extended in vivo investigation, and, most importantly, satellite distribution of the radiotracer to clinical PET centers that do not have radiochemistry facilities. In addition, it shows better imaging characteristics than carbon-11 due to its lower positron energy (0.64 MeV vs. 0.96 MeV for 11C). The larger the positron energy, the larger the average distance the positron travels before annihilating and as such more loss in spatial resolution will be expected. Thus, efforts have been put into developing 18 F-labeled radiotracers. Radiolabeling methods with either carbon-11 or fluorine-18 will not be discussed in this chapter. Several review papers on this topic can be found in the literature (Ametamey et al. 2008; Cai et al. 2008; Miller et al. 2008; Li and Conti 2010).
15.1.3 General Requirements for CNS Radiotracers
The general requirements for successful PET radiotracers for the imaging of specific brain targets such as transporters, receptors, and enzymes also hold true for glutamate receptors. Several excellent review papers have been published on this topic (Waterhouse 2003; Eckelman et al. 2006; Eckelman and Mathis 2006b; Patel and Gibson 2008; Tavares et al. 2012). A summary of some of the most major points is provided below.
15.1.3.1 High Affinity and Selectivity
The affinity of the radiotracer relative to the concentration of binding site present in the target tissue is an important characteristic of site-specific radiotracers (Eckelman et al. 2006). The ability to image a receptor is determined by delivery, by retention at the specific binding site, and by washout of nonspecifically associated radiotracer. Two terms are related to the affinity of a ligand towards its target, and depending on how the value is obtained, the K d (equilibrium dissociation constant) and the K i (inhibition constant) express the affinity of a ligand to the target. Usually, binding affinities should be in the low nanomolar to subnanomolar range. With regard to selectivity, the radioligand in question should have the highest affinity for the actual target and only low affinity for any other binding site.
15.1.3.2 Concentration of Target Sites (B max)
A high ratio of binding site concentration towards radiotracer concentration is a requirement for a valid response to changes in binding site concentration caused by diseases or drug occupancy during a PET experiment. Therefore, it is important that the B max value exceeds the affinity (K d) of the radioligand, and this positively affects the target-to-nontarget ratio and the imaging quality. It is considered that PET tracers with an in vitro B max/K d ratio (also called binding potential) of at least ten have a high chance of providing a specific signal in vivo. A high level of nondisplaceable binding in vivo may require an in vitro B max/K d much greater than ten to provide a useful specific signal in vivo (Eckelman and Mathis 2006b).
15.1.3.3 BBB Permeability
Lipophilicity is a measure of how much of a compound partitions into a lipid environment compared to an aqueous environment. CNS compounds have to pass the blood-brain barrier (BBB) and ideally should therefore have logP values well above 1 to allow free diffusion across the BBB. Nonspecific binding is known to be higher with increasing lipophilicity, and high nonspecific binding in target tissues reduces the signal-to-noise ratio, and thus, the ability to detect the specific signal (Pike 2009). It has been recommended that the logP values of potential CNS compounds should be between 1 and 3.5 (Clark 2003; Waterhouse 2003). It should also be pointed out that lipophilicity values (normally logP or logD) are highly variable, depending on the methodology used for their determination. Values obtained using the traditional “shake-flask” method tends to be lower than those obtained by the HPLC, C18, and calculation methods. LogP values determined using in silico methodology are also highly variable, depending on the algorithm used. Therefore, it has been suggested that logP should not be relied upon as a sole predictor of BBB penetration during brain radiotracer discovery. HPLC measurement of permeability, percentage of plasma protein binding, and membrane interactions may be potentially useful in predicting a radioligand’s in vivo performance (Tavares et al. 2012). Additionally, the BBB possesses efflux pumps which can prevent compounds from effectively accumulating in the brain, of which P-glycoprotein (P-gp) is a key efflux pump (Ohe et al. 2003). Therefore, a PET tracer should not be a substrate for P-gp. There are in vitro assays to determine whether a compound is a P-gp substrate or not (Horio et al. 1989; Braun et al. 2000; Wanger-Baumann et al. 2011).
15.1.3.4 In Vivo Stability
The formation of radioactive metabolites which might enter into the brain and confound the brain signals is undesired. Unfavorable is also the metabolic elimination of the radioactive nuclide from the radiotracer. Thus, the radioligand should be resistant to unfavorable metabolism for the acquisition time, and radioactive metabolites should not be taken up in the target area.
15.1.3.5 Efficient Radiosynthesis
Due to the short half-lives of common PET nuclides, a rapid and efficient radiolabeling process is crucial. The best radiosynthetic scheme should incorporate the radionuclide at the last synthetic step and occur on a time scale similar to the half-life of the radioisotope. Typically, the reaction time for synthetic schemes with multiple steps should be accomplished within two half-lives of the radioisotope. The establishment of a one-step radiosynthesis is desired for obtaining high radiochemical yields with high specific activities. The ratio between labeled and unlabeled tracer is defined by the specific activity. Low specific activities result in low-quality PET images especially for saturable systems such as enzymes, transporter proteins, and receptors. For receptors with low binding sites (low B max), very high specific activity is required in order to exclude substantial occupation of target sites by unlabeled ligand and the saturation of the biological system of interest.
It should be stressed that these criteria do not necessarily guarantee a successful glutamate radiotracer. They are merely guidelines to assist in the selection of potentially useful CNS imaging ligands. Recently, multiparameter optimization methods have been used to assess ligands which have central nervous drug-like properties. The parameters are (1) lipophilicity, calculated distribution coefficient (ClogP); (2) calculated distribution coefficient at pH = 7.4 (ClogD); (3) molecular weight (MW); (4) topological polar surface area (TPSA); (5) number of hydrogen bond donors (HBD); and (6) pK a (Wager et al. 2010, 2011). It is obvious that this multiparameter optimization might also find utility for developing successful CNS PET tracers.
When a potential radiotracer has been selected and a radiolabeling method has been developed, some preclinical evaluations and acute toxicity studies have to be performed prior to PET studies in humans. The characterization of a new radioligand includes autoradiography, biodistribution, and metabolite studies as well as pretreatment and displacement studies, using in vitro, in vivo, and ex vivo techniques. With the increasing ability to carry out PET studies in rodents, using dedicated small animal PET cameras, the evaluation of PET tracers is simplified and serves as an important bridge between laboratory and clinical science (Myers 2001; Lancelot and Zimmer 2010; Xi et al. 2011).
15.2 MGluR1 PET Tracers
MGluR1 has been implicated in neuroprotection, chronic and neuropathic pain, multiple sclerosis, motor dysfunction, epilepsy, cerebral ischemia, and cerebellar long-term depression (Neugebauer 2002; Swanson et al. 2005; Schkeryantz et al. 2007; Wu et al. 2007; Kohara et al. 2008; Ito et al. 2009). Hence, monitoring mGluR1 in vivo and noninvasively would lead to a better understanding of the role of this receptor in the pathophysiology of biological processes mediated through glutamatergic pathways. It has been reported that the highest expression of mGluR1 in rat brain is in the cerebellum; moderate or low level in the thalamus, striatum, hippocampus, and cerebral cortex; and very low level in the brain stem (Fotuhi et al. 1993; Lavreysen et al. 2004a). Since noncompetitive ligands do not bind to the evolutionarily conserved glutamate binding site, they offer the potential for improved selectivity for mGluR subtypes (Parmentier et al. 2000). Therefore, potent and selective allosteric mGluR1 antagonists such as CPCCOEt (Annoura et al. 1996), JNJ-16259685 (Lavreysen et al. 2004b), YM-298198 (Kohara et al. 2005), FTIDC (Suzuki et al. 2007), and MK-5435 (Ito et al. 2009) have been developed (Fig. 15.2). Subsequently, several radiolabeled ligands for PET studies were synthesized and evaluated for their utility as potential imaging agents for the mGluR1 (Fig. 15.3).
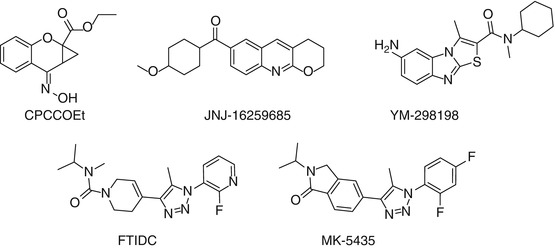
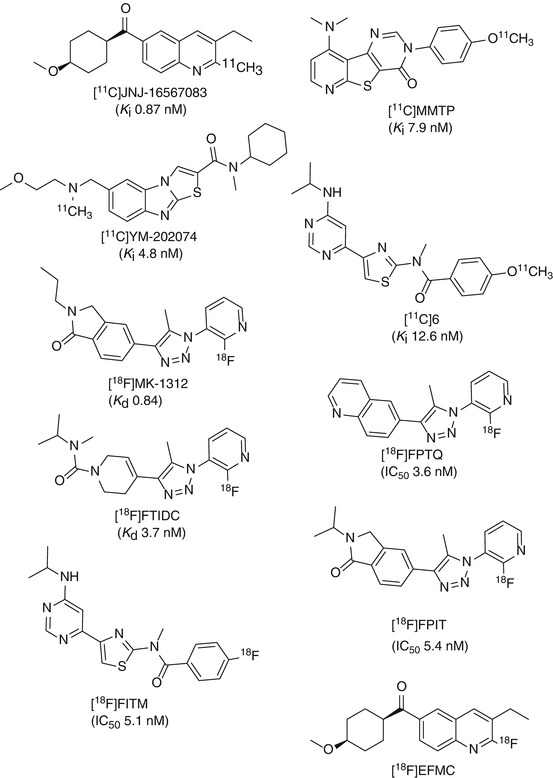
Fig. 15.2
Structures of some mGluR1 allosteric antagonists
Fig. 15.3
Structures of some important mGluR1 PET tracers
15.2.1 11C-Labeled mGluR1 Tracers
The most important carbon-11 radioligands for mGluR1 are described below.
(3-ethyl-2-[11C]-methyl-6-quinolinyl)(cis-4-methoxycyclohexyl)-methanone, also denoted as [11C]JNJ-16567083, was the first reported allosteric antagonist PET tracer for imaging mGluR1 in the rodent brain (Huang et al. 2005). High binding affinity of JNJ-16567083 towards rat mGluR1 receptor was discovered with a K i value of 0.87 nM. [11C]JNJ-16567083 was synthesized via palladium-mediated Stille coupling of the trialkyltin precursor with [11C] methyl iodide (Scheme 15.1). Biodistribution studies in rats under baseline and blockade conditions showed that [11C]JNJ-16567083 is selective and binds specifically to the mGluR1. Among the brain regions examined, the cerebellum, a region with the highest expression of the mGluR1, exhibited the highest uptake of radioactivity and highest specific binding of 81 %. Since the publication of Huang et al., no further evaluation of this radioligand in higher species has been reported.
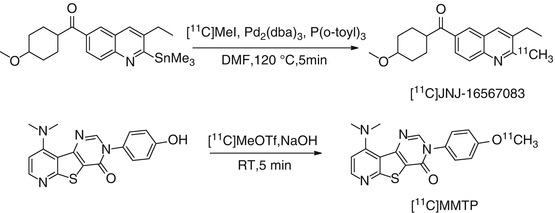
Scheme 15.1
Radiosyntheses of [11C]JNJ-16567083 and [11C]MMTP
[11C]MMTP, [O-methyl-11C]dimethylamino-3(4-methoxyphenyl)-3H-pyrido[3′,2′:4,5] thieno[3,2-d]pyrimidin-4-one, is another promising carbon-11-labeled mGluR1 PET radioligand, which was synthesized by methylation of the corresponding desmethyl precursor using [11C]MeOTf in the presence of NaOH at room temperature (Scheme 15.1) (Prabhakaran et al. 2010). [11C]MMTP selectively labeled mGluR1 binding sites in postmortem human brain slices containing the cerebellum, hippocampus, frontal cortex, and striatum using phosphor imaging autoradiography. The densities of binding sites (B max) for the various brain regions estimated from these autoradiography studies were 140, 110, 85, and 20 fmol/cm2 for cerebellum, hippocampus, frontal cortex, and striatum, respectively. The authors performed PET studies in a baboon and showed that [11C]MMTP penetrates the BBB and accumulated in mGluR1-rich regions. The highest uptake was observed in the cerebellum, a region known to have the highest level of mGluR1 expression.
N-cyclohexyl-6-{[N-(2-methoxyethyl)-N-methylamino]methyl-N-methylthiazolo[3,2-a]benzimidazole-2-carboxamide (YM-202074) is a derivative of YM-298198 (Fig. 15.2) and exhibits an inhibition constant (K i) of 4.8 nM towards mGluR1 and no inhibition up to 1 μM for mGluR5 (Kohara et al. 2008). [11C]YM-202074 was synthesized by N-[11C] methylation of its desmethyl precursor with [11C] methyl iodide in DMF at 90 °C for 3 min. [11C]YM-202074 exhibited optimal lipophilicity with a logD value of 2.7. High specific binding of [11C]YM-202074 for mGluR1 in rat brain was confirmed by in vitro autoradiography under blockade conditions with nonradioactive mGluR1 antagonists YM-202074, JNJ-16259685, and CPCCOEt. However, ex vivo autoradiography and PET study showed that the in vivo radioactivity distribution of [11C]YM-202074 in rat brain region did not reflect the distribution pattern expected from the in vitro autoradiographic results. It was suggested that the discrepancy between in vitro and in vivo distributions might be caused by radiometabolite(s) of [11C]YM-202074 in the brain, and less than 10 % intact compound was found in mouse plasma at 30 min p.i. Therefore, it was concluded that [11C]YM-202074 may not be a useful PET ligand for the in vivo imaging of mGluR1 (Yanamoto et al. 2010).
N-(4-(6-(isopropylamino)pyrimidin-4-yl)-1,3-(thiazol-2-yl)-4-[11C]methoxy-N-methylbenzamide ([11C]6, Fig. 15.3) was prepared by reacting the corresponding phenolic precursor with [11C]MeI (Fujinaga et al. 2012b). A K i value of 12.6 nM was determined towards mGluR1 using rat brain homogenates in competition binding experiments. The logD value of [11C]6 was 2.57 using shake-flask method in octanol/phosphate buffer at pH 7.4. In vitro autoradiography on sagittal section of rat brain slices showed heterogeneous distribution of [11C]6, with highest uptake in the cerebellum, moderate radioactivity in the thalamus, and low in the striatum. The lowest radioactivity was detected in the brain stem. The in vivo PET results confirmed the expected distribution pattern of [11C]6 in rat brain with highest uptake in the cerebellum, followed by thalamus and striatum. Pretreatment with unlabeled 6 (1 mg/kg) significantly reduced the uptake compared to the control group, and the distribution of radioactivity became fairly uniform throughout the brain. The maximum reduction of radioactivity uptake exceeded 85 % in the thalamus and striatum, demonstrating that [11C]6 binds specifically to mGluR1 in the rat brain. The specificity of binding of [11C]6 was also confirmed in mGluR1-knockout mouse brain. Although rapid metabolism was observed in the plasma, > 80 % total radioactivity in the rat brain homogenate was intact [11C]6 (Fujinaga et al. 2012b). Further (pre)clinical evaluation of this promising radioligand is warranted.
15.2.2 18F-Labeled mGluR1 Tracers
A number of fluorine-18-labeled compounds have been evaluated as potential PET ligands for imaging mGluR1. Some of the most important 18F-labeled mGluR1 radioligands reported so far are described below.
15.2.2.1 18F-Labeled Triazole Analogues
Based on the structure of MK-5435, a potent and selective mGluR1 allosteric antagonist (Ito et al. 2009), several pyridyl triazole derivatives have been developed for imaging mGluR1. Depicted in Fig. 15.3 are the structures of four triazole analogues [18F]MK-1312, [18F]FPTQ, [18F]FTIDC, and [18F]FPIT.
[18F]MK-1312 was synthesized via reaction of [18F]KF with the corresponding chloropyridine in DMSO with microwave heating (Scheme 15.2) (Hostetler et al. 2011). Saturation binding studies in cerebellum homogenates with [18F]MK-1312 revealed a K d value of 0.40 and 0.84 nM for rhesus monkey and human cerebellum, respectively. Autoradiography was performed with both human and monkey brain slices. The cerebellum exhibited the highest accumulation of activity for both species. Baseline PET studies in a monkey showed that this radiotracer rapidly penetrates the BBB, which is in line with its optimal lipophilicity value of 2.3. The distribution of [18F]MK-1312 was heterogeneous, with highest uptake observed in the cerebellum and moderate uptake in the thalamus and striatum. Displacement studies with an mGluR1 allosteric antagonist, MK-5435, indicated specific binding of [18F]MK-1312 to mGluR1 in the cerebellum of rhesus monkey, and blockade was concentration dependent (Hostetler et al. 2011). Measurement of in vivo tracer metabolism in plasma revealed that 17 % of plasma radioactivity was due to [18F]MK-1312 at the end of a 90-min baseline PET scan. [18F]MK-1312 possesses the desired characteristics for a potential mGluR1 PET tracer with respect to its lipophilicity, affinity, and specificity towards mGluR1.
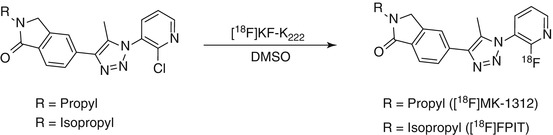
Scheme 15.2
Radiosynthetic scheme for [18F]MK-1312 and [18F]FPIT
[18F]FPTQ, 6-[1-(2-[18F]fluoro-3-pyridyl)-5-methyl-1H-1,2,3-triazol-4-yl]quinolone, was synthesized by heating the bromo precursor with [18F]KF in anhydrous DMSO at 150 °C for 10 min. High binding affinities (IC50 values) of FPTQ towards mouse and human mGluR1 of 1.4 and 3.6 nM, respectively, have been reported (Suzuki et al. 2009). In vitro autoradiographic results showed that the distribution pattern of [18F]FPTQ in the brain of rats was consistent with the distribution of mGluR1 reported previously (Lavreysen et al. 2004a). The specific binding of [18F]FPTQ accounted for higher than 97 % of total binding, as determined by incubating with unlabeled FPTQ or mGluR1-selective JNJ-16259685 in rat brain slices. PET imaging showed that the distribution and uptake of [18F]FPTQ in the rat brain was consistent with the results obtained by dissection as well as by in vitro autoradiography. The maximum standard uptake values (SUV) were 2.3 in the cerebellum, 2.1 in the thalamus, and ranged from 1.1 to 1.5 in the hippocampus, striatum, cerebral cortex, and medulla. However, the presence of a radiolabeled metabolite in the brain may confound the specific binding of this radioligand in vivo. Thus, [18F]FPTQ may be of limited utility for the in vivo investigation of mGluR1 (Fujinaga et al. 2011).
[18F]FTIDC, [4-[1-(2-[18F]fluoropyridine-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl-3,6-dihydropyridine-1(2H)-carboxamide], was labeled with [18F]fluoride by a halogen exchange reaction with the corresponding bromine derivative. Compound FTIDC was identified as a potent and selective mGluR1 allosteric antagonist (Suzuki et al. 2007). It showed high selectivity and equal inhibitory activity towards recombinant human, rat, and mouse mGluR1. The tritiated mGluR1-selective allosteric antagonist, [3H]FTIDC, was identified as a radioligand having high affinity for mGluR1-expressing CHO cells (K d = 2.1 nM) and mouse cerebellum (K d = 3.7 nM) (Suzuki et al. 2009). Brain uptake of [18F]FTIDC in mice was high. The highest accumulation was in the mGluR1‐rich cerebellum at 10 min postinjection and was about two to three times higher than any other brain regions (Ohgami et al. 2009). Although the preliminary studies were promising, no further results have been reported.
[18F]FPIT, 1-(2-[18F]fluoro-3-pyridyl)-4-(2-isopropyl-1-oxoisoindoline-5-yl)-5-methyl-1H-1,2,3-triazole, an analogue of [18F]MK-1312, was synthesized by reacting the bromo precursor with [18F]KF in anhydrous DMSO at 150 °C for 10 min (Scheme 15.2). FPIT was shown to have an IC50 value of 5.4 nM for mGluR1 and 2,500 nM for mGluR5 (Hostetler et al. 2011). The experimentally determined logD value of [18F]FPIT was 2.53, suggesting good BBB penetration. Autoradiography showed that the binding of [18 F]FPIT is consistent with the reported distribution of mGluR1 with high specific binding in the cerebellum and thalamus of rat and monkey brains. [18 F]FPIT displayed a similar distribution of radioactivity in both monkey and rat brains. The sequence of radioactivity level in the brain regions was cerebellum >thalamus >hippocampus >cerebral cortex >striatum >pons-medulla. Pretreatment with FPIT or JNJ-16259865 showed substantial inhibition of [18F]FPIT binding in these brain regions to a level close to or lower than that in the pons-medulla and almost diminished the difference in radioactivity among all brain regions. As indicated by the authors, a disadvantage of [18F]FPIT is its slow kinetics in the monkey brain and also the generation of a radiolabeled metabolite in the brain that could hamper accurate modeling and quantification with PET (Fujinaga et al. 2012a).
15.2.2.2 [18 F]FITM
[18F]FITM (4-[18F]fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide) has a novel chemical structure that does not contain a triazole ring. Moreover, instead of the 2-[18F]fluoropyridine ring, [18F]FITM contains a [18F]fluorobenzene ring, which is expected to be stable against defluorination. [18F]FITM was synthesized by [18F]fluorination of its corresponding nitro precursor with [18F]KF in the presence of Kryptofix 222 (Scheme 15.3). FITM was shown to have potent antagonistic activity against human mGluR1 with an IC50 value of 5.1 nM. Excellent selectivity over other subtypes has been reported whereby the IC50 values were 7,000 nM for human mGluR5, >10,000 nM for mGluR2, and >10,000 nM for mGluR8 (Satoh et al. 2009). The distribution coefficient (logD) of [18F]FITM was 1.46 using the shake-flask method at pH 7.4. Strong signals of radioactivity were found in known mGluR1-rich regions such as the cerebellum and thalamus in in vitro autoradiographic studies using rat brain sections. By co-incubation with the nonradioactive reference FITM or mGluR1 antagonist JNJ-16259685, radioactivity throughout the brain section decreased significantly compared with those in the control section. Upon co-incubation with MPEP, an antagonist for mGluR5, no change in radioactivity accumulation was observed. The signals of radioactivity in ex vivo autoradiography in the brain sections of rat co-injected with JNJ-16259685 decreased by 86–91 % in mGluR1-rich regions compared with those of the control, indicating the tracer was specifically bound to mGluR1 (Yamasaki et al. 2011). Very recently, the same research group reported on the further evaluation of [18F]FITM in a monkey brain. High in vivo stability was also observed. The uptake of [18F]FITM in the monkey brain was not only high in mGluR1-rich cerebellum but also in brain regions such as the thalamus and hippocampus which have moderate expression levels of mGluR1. Specific binding of [18F]FITM could be demonstrated in all the aforementioned brain regions by pretreatment with JNJ-16259685 (Yamasaki et al. 2012).
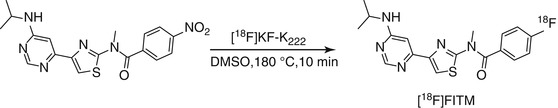
Scheme 15.3
Radiosynthetic scheme for [18F]FITM
15.2.2.3 [18F]EFMC
(3-Ethyl-2-[18 F]fluoroquinolin-6-yl) cis-(4-methoxycyclohexyl)methanone ([18 F]EFMC), the 18F-labeled analogue of JNJ-16567083, was prepared by Br–18F exchange in DMSO in the presence of Kryptofix 222 at 100 °C for 5 min, followed by semi-HPLC purification (Lee et al. 2012). The radiochemical yield of [18F]EFMC was 29 %. No degradation of [18F]EFMC was found in MeCN/H2O (1:1) after 4 h at room temperature, and 90 % intact compound was present in human serum after 2 h of incubation at 37 °C. In biodistribution studies, the brain uptake of [18F]EFMC was found to be 4.35 ± 0.32 % ID/g at 10 min after injection. Highest uptake of [18F]EFMC was observed in the cerebellum in a PET study. Pretreatment with unlabeled EFMC led to a remarkable decrease of cerebellum uptake indicating specific binding towards mGluR1 in mice brain.
So far, mGluR1 PET ligands such as [18F]FTIDC, [18F]MK-1312, [18F]FPIT, and [18F]FPTQ containing a triazole and a 2-[18F]fluoropyridine ring have shown high specific binding to mGluR1-rich regions in rodent and primate brains in vitro and in vivo. For example, [18F]FPIT had about fivefold higher SUV value than [11C]YM-202074 (Yanamoto et al. 2010) and 0.5-fold lower uptake (SUV) than [11C]JNJ-16567083 (Huang et al. 2005) in the rat cerebellum. However, to the best of our knowledge, none of these radioligands has progressed into a clinical setting. Based on the currently available results, it is suggested that radioligands containing a triazole and 2-[18F]fluoropyridine ring are metabolized through similar in vivo elimination systems, and therefore ligands of this structural type may not be suitable for in vivo quantitative analysis of mGluR1. Among all the mGluR1 ligands evaluated so far, [18F]FITM, a pyrimidine analogue, appears to show the most promising in vivo characteristics due to its higher metabolic stability and in vivo specificity. Thus, the expectations are that [18F]FITM will soon find application in the clinic as a useful PET ligand for determining the distribution of mGluR1 expression in humans and also to ascertain the density of mGluR1 in pathological states in which mGluR1 is implicated.
15.3 MGluR5 PET Tracers
MGluR5 is an important target for PET imaging with potential for applications in drug development, diagnosis, and therapy monitoring. Metabotropic glutamate subtype 5 receptor (mGluR5) was cloned in 1992 and exists as a covalently bound homodimer in heterologous expression system as well as in native tissue (Abe et al. 1992; Romano et al. 1996). It is expressed in brain regions such as the hippocampus, basal ganglia (striatum and nucleus accumbens), amygdala, and cortex (Romano et al. 1995; Gupta et al. 2005). MGluR5 has been implicated in a number of central nervous system disorders including depression, anxiety, addiction, schizophrenia, Parkinson’s disease, and fragile X syndrome (Ohnuma et al. 1998; Rouse et al. 2000; Spooren et al. 2000, 2003; Tatarczynska et al. 2001; Spooren and Gasparini 2004; Bear 2005; Palucha et al. 2005; Tsai et al. 2005; Pietraszek et al. 2007; Carroll 2008). As such, this receptor provides a novel target for the development of therapeutic drugs for the aforementioned neurological disorders. A number of noncompetitive mGluR5 ligands have been developed during the past years, and their potential as therapeutic drugs for some of the abovementioned CNS disorders has been investigated (Varney et al. 2002; Augelli-Szafran and Schwarz 2003; Chua et al. 2005; Porter et al. 2005; Szydlowska et al. 2007; Carroll 2008; Chen and Conn 2008; Gasparini et al. 2008; Jaeschke et al. 2008; Kulkarni et al. 2009; Lindsley and Emmitte 2009; Ritzen et al. 2009; Emmitte 2011; Lindemann et al. 2011). In addition to CNS disorders, mGluR5 was found to be upregulated in CNS tumors and some non-CNS tumors (Brocke et al. 2010; Choi et al. 2011), participating in tumor growth and aggression.
The first potent and selective noncompetitive mGluR5 antagonists reported were the diaryl alkynes, 2-methyl-6-(phenylethynyl)pyridine (MPEP) (Gasparini et al. 1999; Lea and Faden 2006), and 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) (Fig. 15.4) (Cosford et al. 2002, 2003a, b). Since their publication, MPEP and MTEP have been used as templates for the design of noncompetitive mGluR5 antagonists. Also a series of PET radioligands based on their core structures have been labeled with either carbon-11 or fluorine-18 and evaluated in in vitro and in vivo studies. Two reviews providing an overview on available mGluR5 PET ligands have recently been published (Yu 2007; Mu et al. 2010).
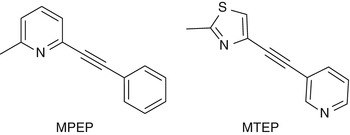
Fig. 15.4
Chemical structures of MPEP and MTEP
15.3.1 MPEP-Derived mGluR5 Tracers
MPEP and its methyl analogue M-MPEP have been identified as potent, highly selective, noncompetitive antagonists for mGluR5. Derivatives of MPEP that have been labeled with either carbon-11 or fluorine-18 are depicted in Fig. 15.5.
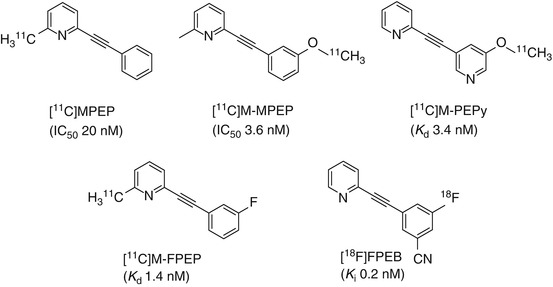
Fig. 15.5
Chemical structures of 11C- and 18F-labeled MPEP derivatives
The high binding affinity of MPEP (IC50 20 nM) (Gasparini et al. 1999, 2002) to the heptahelical domain mGluR5 initiated the first radiolabeling of this compound with tritium. The identification of high-affinity analogues of M-MPEP (2-(2-(3-methoxyphenyl)ethynyl)pyridine, IC50 3.6 nM) (Gasparini et al. 2002) and M-PEPy (3-methoxy-5-pyridin-2ylethynyl)pyridine, K d 3.4 nM) (Anderson et al. 2003; Cosford et al. 2003a; Patel et al. 2003) prompted the radiolabeling of these derivatives with carbon-11 for evaluation as potential candidates for imaging mGluR5.
[11C]MPEP was prepared via Stille coupling reaction of 2-(phenylethynyl)-6-(trimethylstannyl)pyridine with [11C]MeI (Scheme 15.4) in high radiochemical purity and good specific radioactivity (33–44 GBq/μmol) (Yu et al. 2005). In vivo PET imaging studies of [11C]MPEP in Sprague–Dawley rats showed high accumulation of [11C]MPEP in brain areas such as the olfactory bulb, striatum, hippocampus, frontal cortex, and cerebellum. Pre-administration of unlabeled MPEP (10 mg/kg) 5 min prior to administration of [11C]MPEP led to a decreased [11C]MPEP binding in the olfactory bulb. In contrast, an increased uptake was noted for the hippocampus and striatum suggesting that [11C]MPEP binds nonspecifically to these brain regions. No reasons have been provided by the authors to explain this phenomenon. For the olfactory bulb, the extent of specific binding was 45 and 61 % at 5 and 40 min postinjection, respectively.
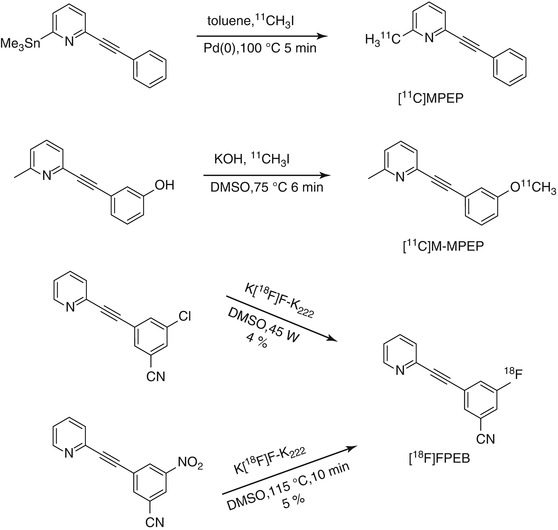
Scheme 15.4
Radiosyntheses of [11C]MPEP, [11C]M-MPEP, and [18F]FPEB
The radiosynthesis of [11C]M-MPEP was accomplished by reacting [11C]MeI with phenolic precursor in the presence of solid KOH powder in 85 % radiochemical yield and 98 % radiochemical purity (Scheme 15.4) (Yu et al. 2005). Moderate uptake was observed after tail vein injection of [11C]M-MPEP into rats. Pre-administration of MPEP (10 mg/kg) induced a 60 % decrease in [11C]M-MPEP binding in the olfactory bulb 5 min postinjection. In other brain regions, the mGluR5 antagonist, MPEP, decreased [11C]M-MPEP binding only during the early time points: 45 % in the cortex, 14 % in the hippocampus, and 29 % in the striatum at 5 min postinjection. At 40 min postinjection, a binding ratio of olfactory bulb to cerebellum was 4.2. Kokic et al. also reported on the radiosynthesis and the in vivo evaluation of [11C]M-MPEP albeit in an abstract (Kokic et al. 2001). In their study, Kokic and colleagues found an increased uptake of [11C]M-MPEP radioactivity 30 min postinjection in the rat brain after co-injection with unlabeled M-MPEP (1 mg/kg). However, no information was provided on radioactivity uptake in the olfactory bulb.
[11C]M-PEPy was obtained by reacting the desmethyl precursor with either [11C]MeI (Yu et al. 2005) or [11C]CH3OTf (Severance et al. 2006) in the presence of KOH or NaOH in moderate radiochemical yield. In vitro phosphor imaging with [11C]M-PEPy using human and rat brain tissues demonstrated high specific binding in the hippocampus, striatum, and cortex with minimal specific binding in the cerebellum. The hippocampus-to-cerebellum ratio found in this study was 37 in rats and 8 in humans. However, further in vivo micro-PET studies in rats using urethane anesthesia, PET studies in baboons using isoflurane anesthesia, and ex vivo studies in rats showed little specific binding in the brain and very fast tracer washout (Severance et al. 2006). One possible explanation is that [11C]M-PEPy is actively extruded from the brain by P-gp or some other multidrug-resistant like transport protein in the blood-brain barrier. Despite the promising in vitro results, the low signal-to-noise ratio found in vivo did not justify the use of [11C]M-PEPy as a PET radiotracer in humans (Severance et al. 2006).
Kessler labeled 2-methyl-6-(3-fluoro-phenylethynyl)-pyridine (M-FPEP), another analogue of MPEP, with 11C (Kessler 2004). [11C]M-FPEP was prepared in good radiochemical yields and high specific radioactivity starting from its bromo-pyridine precursor. Saturation assays of [11C]M-FPEP binding resulted in a single high-affinity binding site with a K d of 1.4 ± 0.1 nM and a B max value of 563 ± 190 fmol/mg protein. Dynamic PET studies in a rat indicated rapid uptake of [11C]M-FPEP in rat brain, followed by a fast clearance. Classical biodistribution studies in rats showed a homogeneous distribution of [11C]M-FPEP in all brain regions examined, suggesting nonspecific uptake in the brain, which was also confirmed in blocking studies.
Based on the data summarized in Table 15.1, although moderate to high binding affinities towards mGluR5 for all the 11C-labeled MPEP derivatives have been observed, their utility as mGluR5 imaging agents has been limited due to their unfavorable in vivo properties. The radiosynthesis of [18F]FPEB ([18F]3-fluoro-5-[(pyridin-3-yl)ethynyl] benzonitrile), a structural analogue of M-FPEP, was accomplished via a nucleophilic aromatic substitution reaction using either nitro or Cl− as a leaving group under conventional or microwave heating conditions (Hamill et al. 2005; Patel et al. 2007; Wang et al. 2007; Belanger et al. 2008). Low radiochemical yields were obtained under both conditions (Scheme 15.4). The low radiochemical yields may be due to the position of the leaving group which is not activated enough for aromatic nucleophilic substitution. High binding affinities of [18F]FPEB towards mGluR5 in rat, rhesus, and human brain sections have been reported (K d = 0.1–0.15 nM, n ≥ 3) (Patel et al. 2007). [18F]FPEB showed an approximately 70 % specific signal in the cortex and caudate-putamen of rat, rhesus, and human brain (Patel et al. 2007). Small animal PET imaging studies using Sprague–Dawley rats demonstrated that [18F]FPEB accumulated in mGluR5-rich regions of the brain such as striatum and hippocampus with 77 % specific binding after 20 min postinjection (Patel et al. 2005). In blocking studies of [18F]FPEB, pretreatment with 3 mg/kg, MTEP did not result in identical residual uptake in all regions. When [18F]FPEB was blocked with 10 mg/kg MTEP, nearly homogeneous uptake of tracer in all regions was observed, indicating full blockade had been achieved. It was also found that blocking mGluR5 is more efficient with MTEP than MPEP under the same conditions (Wang et al. 2007). PET imaging studies in a monkey also showed that [18F]FPEB readily enters the brain and provides mGluR5-specific signal in all gray matter regions, including the cerebellum (Hamill et al. 2005). Initial human studies have been reported by Tamagnan et al. (2009). Dynamic PET images of six human subjects examined so far revealed excellent penetration of [18F]FPEB into human brain with initial visualization of anterior cingulate, thalamus, caudate, and midbrain, while cerebellum showed low uptake.
Table 15.1
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Tracers for Beta-Amyloid and Other Proteinopathies
 PET Imaging of ABC Transporters in the BBB
PET Imaging of ABC Transporters in the BBB
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT




Lipophilicity, in vitro binding affinity, and specificity of 11C-labeled MPEP derivatives
Related posts:
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Imaging of ABC Transporters in the BBB
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
