Fig. 18.1
Structure of NMDARs and location of ligand-binding site
Positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging for NMDARs could be powerful tools for obtaining information about the functional mechanism of the NMDA ion channel in the living brain, the pathophysiology of related neurological disorders, and in vivo occupancy of agonists or antagonists of NMDARs. Although a conclusive relationship between NMDAR expression and neurodegenerative disease has yet be elucidated, several reports suggested that NMDAR expression is deregulated in neurological patients. It is reported that mRNA and protein levels of NMDAR NR2A and NR2B subunits were decreased in AD patients (Hynd et al. 2004; Jacob et al. 2007). In addition, [3H]Ro 25–6981 (NR2B selective ligand as described below) binding was increased in the putamen of PD patients experiencing motor complications (Calon et al. 2003), and protein levels of the NR1 subunit were decreased in the postmortem left hippocampus in schizophrenic patients (Vrajová et al. 2010). Therefore, nuclear medicine imaging of NMDARs could lead to characterization of disease progression and early diagnosis of such diseases.
Development of imaging agents for NMDARs has been extensively reviewed previously (Waterhouse 2003a; Sobrio et al. 2010). General strategies for the development of radioligands for NMDARs have been based on the structural modification of NMDAR antagonists for application in PET or SPECT imaging. Thus, the target sites for the radioligands could be divided into three classes as follows: (1) PCP-binding site (channel blocker-binding site), (2) glycine-binding site, and (3) ifenprodil-binding site (NR2B negative modulator binding site). Because most competitive antagonists for the glutamate-binding site have an acid moiety that may cause low brain uptake, no radioligands for this site have been published in the literature. Despite enormous efforts, there are still no satisfactory radioligands for the visualization of NMDARs in the living brain. In this chapter, we summarize the history and present status of radioligands for these three sites.
18.2 Radioligands for PCP-Binding Site
Open channel blockers of NMDARs, such as MK-801 and PCP derivatives (Fig. 18.2), have been reported to bind to NMDARs in a receptor activation-dependent manner (Loo et al. 1986; Foster and Wong 1987; Wong et al. 1988). Thus, in vivo imaging of the PCP-binding site of NMDARs would provide not only quantification of NMDAR density in the living brain but also allow receptor activation under normal and pathophysiological conditions to be monitored. In recent decades, a number of reports have emerged regarding the development of radioligands based on dissociative anesthetics (PCP, MK-801, ketamine, memantine derivatives, diarylguanidines) as shown in Fig. 18.2.
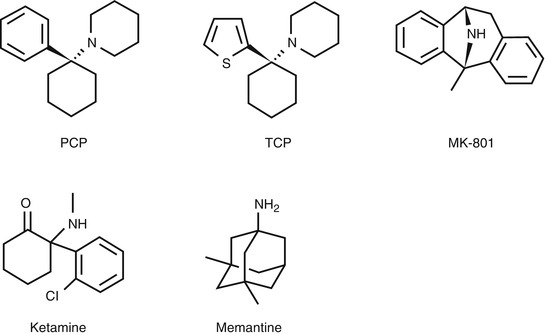
Fig. 18.2
Chemical structure of open channel blockers of NMDARs
18.2.1 Dissociative Anesthetic Derivatives
18.2.1.1 PCP and TCP Derivatives
PCP, one of the dissociative anesthetics, has been found to inhibit the actions of NMDARs by blocking the ion channel (Anis et al. 1983). On the other hand, TCP ([2-thienyl]-cyclohexyl-PCP), one of the PCP derivatives, has been reported to show severalfold higher affinity than PCP itself toward the PCP-binding site (Vincent et al. 1979; Tsukiyama et al. 1991).
In structure-activity relationship (SAR) studies of PCP derivatives, the binding affinity of the compounds was expressed as K 0.5, which means the half maximal (50 %) inhibitory concentration of [3H]PCP binding. Methoxy analogs 1 and 2 (Fig. 18.3) showed a higher binding affinity (1; K 0.5 = 90 nM, 2; K 0.5 = 58 nM) than PCP itself (PCP; K 0.5 = 250 nM) (Chaudieu et al. 1989). The IC50 value of methoxymethyl analog of TCP (3, Fig. 18.3) for the [3H]TCP-binding site was 16 nM, which was slightly weaker than TCP (IC50 = 7.9 nM) but stronger than PCP (IC50 = 52 nM) (Tsukiyama et al. 1991). These compounds were labeled with C-11 and then evaluated as PET radioligands for the PCP-binding site of NMDARs (Haradahira et al. 1998). The [11C]-radiotracers were prepared from hydroxyl precursors reacting with [11C]CH3I to reach a specific activity of more than 41 GBq/μmol. In the biodistribution studies in mice, initial uptake of the TCP analog [11C]3 was twofold higher (2.6 − 2.9 % ID/g at 1 min) than the PCP analogs [11C]1 (1.4 − 1.6 % ID/g at 1 min) and [11C]2 (1.1 − 1.5 %ID/g at 1 min). On the other hand, [11C]3 showed faster clearance in the brain than [11C]1 and [11C]2. However, there was no significant difference in the accumulated radioactivity level between the NMDAR-rich regions and NMDAR-poor regions. These results indicated that the radioligands had no small binding interaction with brain components other than PCP-binding site. Since no blocking studies using the PCP-binding site antagonists were performed, it is unclear whether these radioligands bound specifically in certain brain regions (Haradahira et al. 1998).
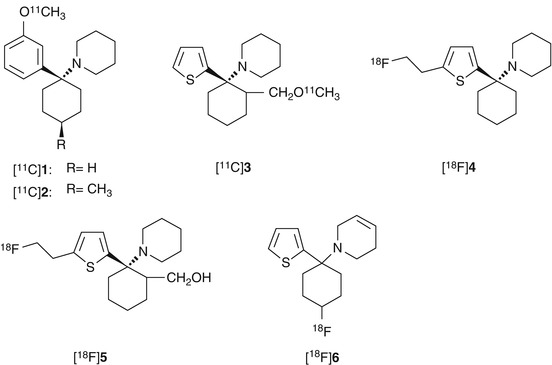
Fig. 18.3
Chemical structure of radiolabeled PCP and TCP derivatives
Orita et al. developed [18F]fluoroethyl analog of TCP ([18F]4, Fig. 18.3) with IC50 values of 61 nM for the PCP-binding site of NMDARs. The [18F]4 showed heterogeneous in vivo distribution that was similar to NMDAR expression. In addition, co-injection of high-affinity ion-channel blocker cis-HPTC (1.7 μmol/kg) yielded reduction of regional cerebral distribution of [18F]4 (Fig. 18.4). However, this tracer may be an unsuitable PET radioligand for NMDARs because of the high nonspecific binding in brain tissues (Orita et al. 1993).
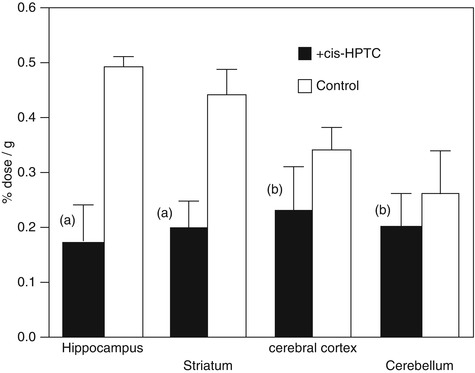
Fig. 18.4
Regional brain distribution of radioactivity in rats at 15 min after injection of [18F]4. In the blocking study, cis-HPTC (1.7 mmol/kg) was pre-injected 5 min before administration of [18F]4. Student’s t-test with respect to the control, (a) P < 0.01, (b) not significant (Orita et al. 1993)
Another [18F]fluoroethyl analog of TCP ([18F]5, Fig. 18.3) has been synthesized by nucleophilic substitution of mesylate precursor with 18F− at a specific activity of >31 GBq/μmol. Biodistribution analysis in rats revealed that [18F]5 exhibited peak uptake (0.25 % ID/g) in the brain at 10 min. However, no regional difference was observed in the brain tissues, and the blocking effect of cis-HPTC (1.7 μmol/kg) was not observed for all the tissues investigated. Nonspecific brain distribution of [18F]5 could be attributed to the hydrophobic property and low affinity for NMDARs (IC50 = 1.5 μM vs [3H]TCP) of these ligands (Shibayama et al. 1996).
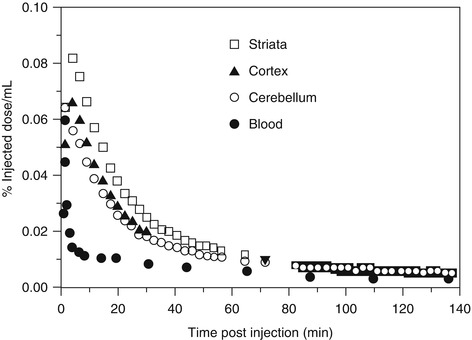
Ouyang et al. has reported a fluorocyclohexyl analog of TCP ([18F]6, Fig. 18.3) with IC50 value of 97 nM for the PCP-binding site (vs [3H]MK-801). The [18F]6 was prepared by nucleophilic substitution of corresponding tosylate precursor with a specific activity of 19–37 GBq/μmol. In PET studies in the rhesus monkey, [18F]6 showed up to 0.08 %ID/mL of brain uptake, but the radioactivity was cleared rapidly from all regions of the brain indicative of little or no retention of the radiotracer in the receptor-rich regions (Fig. 18.5) (Ouyang et al. 1996). Because [18F]6 has only moderate affinity and the specificity for NMDARs has not been confirmed, new radioligands with significant improvement of binding affinity and target specificity have to be developed.
18.2.1.2 MK-801 Derivatives
The anticonvulsant MK-801 (Fig. 18.2) has been identified as a potent and selective noncompetitive NMDAR antagonist (Wong et al. 1986). Because MK-801 has high binding affinity for the PCP-binding site of NMDARs (Wong et al. 1988) and easy penetrability into the brain (Clineschmidt et al. 1982), MK-801 analogs were expected to be suitable as in vivo imaging probes for NMDARs. Several MK-801 derivatives labeled with 11C, 18F, or 123I have been synthesized and evaluated as PET or SPECT radioligands for NMDARs.
Blin et al. reported the preparation of [18F]fluoromethyl analog of MK-801 ([18F]FMM, Fig. 18.6) and PET studies on baboons (Blin et al. 1991). In the baseline PET studies, initial accumulation into the brain of [18F]FMM was marked with peak uptake values as high as 0.03 %ID/mL. However, the ratios of [18F]FMM in the cerebral cortex and striatum (NMDAR-rich regions) to the cerebellum (NMDAR-poor regions) were only minimal (maximal ratios ranging from 1.1 to 1.6 during the first 20–40 min). In addition, there was no clear reduction in the brain uptake and target-to-nontarget ratios by pretreatment of the open channel blockers (PCP or MK-801). Because binding of [3H]MK-801 to the PCP-binding site has been enhanced severalfold in the presence of high amount of glutamate (Wong et al. 1988), the authors investigated further using PET studies of [18F]FMM on ischemic baboons with excessive glutamate activation. However, the brain kinetics of [18F]FMM was not significantly different between the ischemic and control groups. It can be concluded that [18F]FMM is an unsuitable radioligand for the PCP-binding site of NMDARs (Blin et al. 1991). Ransom et al. developed the 3-[123I]iodo analog of MK-801 ([123I]3-iodo-MK-801, Fig. 18.6) as a SPECT imaging agent. Over 95 % of [123I]3-iodo-MK-801 binding was specific in rat brain homogenates. MK-801 derivatives were shown to bind with high affinity for the [123I]3-iodo-MK-801-binding site {3-iodo-MK-801: IC50 = 3.4 nM, (+)-MK-801: IC50 = 5.6 nM} indicating excellent binding affinity of this tracer for the PCP-binding site (Ransom et al. 1990). These positive in vitro results prompted further in vivo imaging studies. Owens et al. reported the clinical SPECT investigation of [123I]3-iodo-MK-801. In normal subjects, [123I]3-iodo-MK-801 showed rapid uptake into the brain, but the tracer has a high nonspecific retention in the brain tissues probably due to its high lipophilicity (Owens et al. 1997). Brown et al. reported the clinical SPECT studies of [123I]3-iodo-MK-801 in Alzheimer’s diseases (AD) and healthy subjects. There were slight difference in the brain uptake/rCBF (regional cerebral blood flow) ratio and wash out of [123I]3-iodo-MK-801 between the AD patients and control groups, which suggested that the binding of this tracer in the brain was increased in AD patients. However, the authors concluded that [123I]3-iodo-MK-801 did not provide accurate measurement of NMDARs since the alteration was not significant, and the brain distribution of this tracer was homogenous in both AD and control groups (Brown et al. 1997).
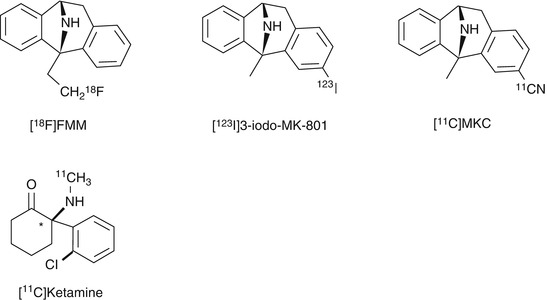
Fig. 18.6
Chemical structure of radiolabeled MK-801 derivatives and [11C]ketamine
Andersson et al. successfully synthesized 3-[11C]cyano analog of MK-801 ([11C]MKC, Fig. 18.6) with high specific activity (220–600 GBq/μmol) from 3-iodo precursor using palladium(0)-promoted [11C]-cyanation (Andersson et al. 1998). Saturation assay in rat cortical membranes revealed that [11C]MKC had a single high-affinity binding site with a K d of 8.2 nM and a B max of 1.6 pmol/mg protein (Sihver et al. 1998). In vitro autoradiography demonstrated that the highest specific binding in rat slices of [11C]MKC was observed in the hippocampus and frontal cortex. Moderate binding was found in the caudate putamen and thalamus. Low-density binding sites were observed in the cerebellum, pons, midbrain, and brainstem (Fig. 18.7). This correlated with the distribution of [3H]MK-801 (Bowery et al. 1988). The in vivo PET studies demonstrated that [11C]MKC showed a rapid and high uptake in the brain tissues. In addition, the radioactivity level was higher in the frontal cortex than cerebellar cortex, but the brain distribution pattern was well correlated with the rCBF (Fig. 18.8). Neither MK801 (0.1 mg/kg) nor ketamine (2.5 mg/kg) as pretreatment dose caused a significant change in regional brain kinetics. These results could be caused by the very low-specific binding fraction of [11C]MKC under in vivo conditions (Sihver et al. 1998). Development of new radioligands with lower lipophilicity and higher NMDAR selectivity should be tried to reduce nonspecific binding.
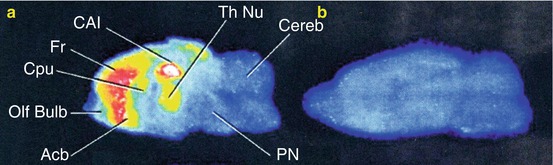
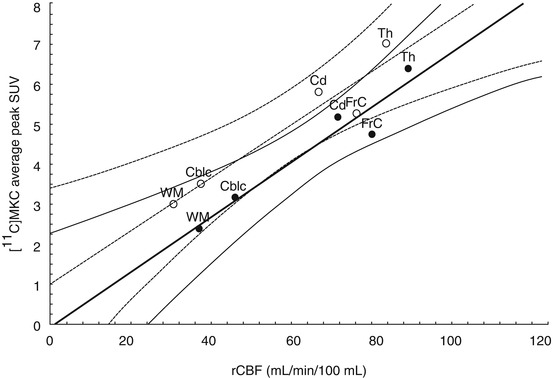
Fig. 18.7
In vitro autoradiography of [11C]MKC in the rat brain slices. Total binding (a) and nonspecific binding (b) are shown in sagittal planes. Nonspecific binding was measured in the presence of MK-801 (1 mM). Abbreviations: Acb accumbens nucleus, CA1 CA1 of hippocampus, Cereb cerebellum, Cpu caudate putamen, Fr frontal cortex, Olf bulb olfactory bulb, PN pontine nuclei, Th Nu thalamic nuclei (Sihver et al. 1998)
Fig. 18.8
Typical graph presenting the relation between peak [11C]MKC uptake and rCBF in the same regions in the baseline measurement (open circles and dotted lines) and ketamine-pretreatment measurement (closed circles and continuous lines). Curved lines indicate 95 % confidence. Abbreviations: FrC frontal cortex, Cd right caudate nucleus, Th thalamic nuclei, CblC cerebellar cortex, WM white matter (Sihver et al. 1998)
18.2.1.3 [11C]Ketamine
Ketamine (Fig. 18.2) is known as a PCP-like anesthetic drug which has a weak binding affinity for the PCP-binding site of NMDARs (Lodge and Johnson 1990). In the displacement assay using pig brain homogenates, the K i values of (S)-ketamine and (R)-ketamine for [3H]MK-801-binding site of NMDARs were 1.2 and 5.0 μM, respectively (Oye et al. 1992). [11C]-labeled (R,S)-ketamine ([11C]ketamine, Fig. 18.6) and its enantiomers were synthesized, then in vivo evaluation was performed (Hartvig et al. 1994; Shiue et al. 1997). Although [11C]ketamine showed high blood–brain barrier (BBB) permeability (2.80 % ID/g at 5 min after injection) in mice brain, unchanged [11C]ketamine was only 50 % at 5 min. PET studies in a baboon demonstrated that the influx of [11C]ketamine and its enantiomers into the brain was high for the first few minutes, but radioactivity then declined rapidly and a similar uptake in the striatum and cerebellum was observed. In addition, these three tracers were also metabolized rapidly in the baboon plasma. Because [11C]ketamine and its enantiomers were revealed to be metabolically unstable and had inconsistent binding with NMDAR expression, these tracers failed to act as useful radioligands for in vivo imaging of NMDARs (Shiue et al. 1997).
18.2.1.4 Memantine Derivatives
Memantine (Fig. 18.2) is a drug used in the treatment of various neurological and psychiatric disorders, including Parkinson’s disease and Alzheimer’s disease. Memantine has been identified as a noncompetitive open channel blocker of NMDARs with good BBB permeability (Parsons et al. 1993; Lipton 2004). Therefore, several radiolabeled memantine derivatives have been developed as imaging agents for NMDARs.
Synthesis of 3-[18F]fluoromethyl analog of memantine ([18F]MEM, Fig. 18.9) was achieved from N-Boc-protected tosylate precursor by nucleophilic radiofluorination in two steps. The MEM showed a slightly weaker inhibitory activity to the current response to NMDARs (IC50 = 6.0 μM) than memantine (IC50 = 2.9 μM) and PCP (IC50 = 1.0 μM). In the biodistribution studies in mice, [18F]MEM showed an excellent brain uptake (3.6 % ID/g at 60 min) and the accumulation in the frontal cortex and hippocampus was higher than in the cerebellum. In addition, co-injection of MK-801 (0.10 mg/kg) led to 25–36 % reduction of radioactivity in NMDAR-rich regions. PET studies in a rhesus monkey revealed that [18F]MEM displayed high brain uptake and low clearance of the radiotracer from the brain tissues. Pretreatment with memantine (0.50 and 1 mg/kg) yielded a reduction of uptake of up to 32 % from 60 min in the examined brain regions. However, the highest uptake was observed in the cerebellum, which is an NMDAR-poor region. In addition, pretreatment with haloperidol, which is a ligand with high affinity for dopamine D2 and sigma receptors, also caused similar reduction of radioactivity in the brain (Samnick et al. 1998). In clinical PET studies, [18F]MEM showed perfusion-dependent uptake in the brain rather than NMDAR expression (Ametamey et al. 2002). Because of inconsistent binding with NMDAR expression and low specificity for NMDARs, it would appear that [18F]MEM is an unsuitable radioligand (Samnick et al. 1998; Ametamey et al. 2002). However, the different brain distribution pattern of [18F]MEM observed among species was noteworthy. New technetium-labeled memantine derivatives such as [99mTc]NCAM and [99mTc]NHAM have recently been reported as novel SPECT imaging probes (Fig. 18.9). The [99mTc]NCAM had a single binding site with a K d of 701 nM. Furthermore, the in vitro binding of [99mTc]NCAM was blocked by ketamine and MK-801 in a competitive manner. In the biodistribution studies in mice, [99mTc]NCAM showed 1.2 % ID/g brain uptake at 5 min and the target (frontal cortex, hippocampus) to nontarget (cerebellum) ratio of radioactivity reached 2.5 − 3.0 at 60 min. The in vivo blocking studies were not so far reported in the literature (Zhou et al. 2012).
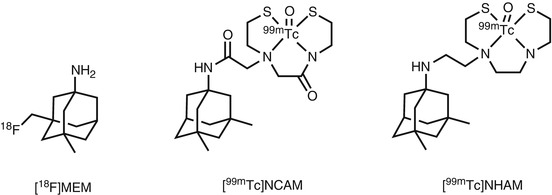
Fig. 18.9
Chemical structure of radiolabeled memantine derivatives
18.2.2 Diarylguanidine Derivatives
Initially reported imaging probes, such as PCP and MK-801 analogs, suffered from high nonspecific in vivo accumulation, which was probably due to the high lipophilicity and lack of selectivity for NMDARs. Diarylguanidine derivatives were discovered as NMDAR open channel blockers with high potency and selectivity. These compounds showed therapeutic effect in various brain disease model animals (Reddy et al. 1994; Hu et al. 1997). Because most diarylguanidines are more hydrophilic than PCP and MK-801, several radiolabeled diarylguanidine analogs were developed for PET or SPECT imaging of NMDARs. In the first study, [125I]CNS 1261 (Fig. 18.10) was developed as a high-affinity imaging agent for NMDARs with a K i value of 4.2 nM for the [3H]MK-801-binding site with moderate lipophilicity (logD = 2.19). Binding assays to other neurotransmitter receptors demonstrated that CNS 1261 had a weaker binding affinity for the other 41 neurotransmitter receptors. Ex vivo autoradiography studies with normal rats showed that the [125I]CNS 1261 uptake in the hippocampus was 2.4–2.9-fold higher than that in the cerebellum, in which the accumulation matched the distribution of NMDARs. In the ischemic rat brain, [125I]CNS 1261 uptake was considerably increased in the neocortex and striatum, with a twofold higher uptake in the ischemic hemisphere of the caudate nucleus uptake than the same regions on the contralateral nonischemic hemisphere (Fig. 18.11). This suggested that [125I]CNS 1261 could recognize the areas of NMDAR activation (Owens et al. 2000). Based on these results, several clinical SPECT studies of [123I]CNS 1261 were conducted. SPECT imaging of [123I]CNS 1261 in healthy volunteers were conducted by using both bolus and bolus plus constant infusion paradigms. Total volume of distribution (V T) values was approximately the same in the NMDAR-rich regions (striatum, hippocampus, frontal cortex) and NMDAR-poor region (cerebellum). The V T value in the thalamus, which is a region with considerably high expression of NMDARs, was slightly higher than in other regions (Fig. 18.12) (Erlandsson et al. 2003; Bressan et al. 2004). Bolus plus infusion of ketamine in healthy volunteers caused a global reduction in V T of [123I]CNS 1261 compared with placebo in most brain regions, including the putamen, temporal, and pericallosal regions (Stone et al. 2006). Subsequent work by the same group demonstrated that the degree of reduction in the V T values of [123I]CNS 1261 correlated with the negative symptoms of schizophrenia in all brain regions examined (Stone et al. 2008). In addition, they reported that drug-free patients with schizophrenia had reduced binding of [123I]CNS 1261 in the left hippocampus relative to the whole cortex compared with healthy controls (Pilowsky et al. 2006). In contrast, the same group found that the V T values of [123I]CNS 1261 did not differ between healthy normal volunteers and drug-free or typical antipsychotic-treated schizophrenia patients. On the other hand, clozapine-treated patients showed a significant reduction of V T in all brain regions examined, which indicated a decrease of NMDAR activation in this group. However, the regional brain distribution of [123I]CNS 1261 was almost homogenous in all groups (Bressan et al. 2005). Although there was no conclusive evidence that [123I]CNS 1261 could recognize NMDAR activation, these reports suggested that protein expression levels of NMDARs could be reduced in the brain of schizophrenia patients. Numerous reports indicated the involvement of NMDAR hypofunction in the pathophysiology of positive and negative symptoms of schizophrenia (Kantrowitz and Javitt 2010; Lin et al. 2012). Recently, Knol et al. reported further in vivo studies of [123I]CNS 1261 using rats. The [123I]CNS 1261 showed higher accumulation in the NMDAR-rich regions such as hippocampus and frontal cortex than that of NMDAR-poor cerebellum, in which the ratio was maximum (about 1.4) at 2 h postinjection. Pretreatment with the NMDAR co-agonist, d-serine, (10 mg/kg) led to no significant change in the hippocampus/cerebellar ratio. On the other hand, treatment with d-serine and MK-801 resulted in a significantly lower hippocampus/cerebellar ratio, which indicated the existence of specific trapping of a certain amount [123I]CNS 1261 in the NMDAR-rich regions. However, the authors concluded that the use of [123I]CNS 1261 would be limited in terms of quantification and detection of small changes in receptor availability because of a considerable amount of nonspecific binding of this tracer (Knol et al. 2009).
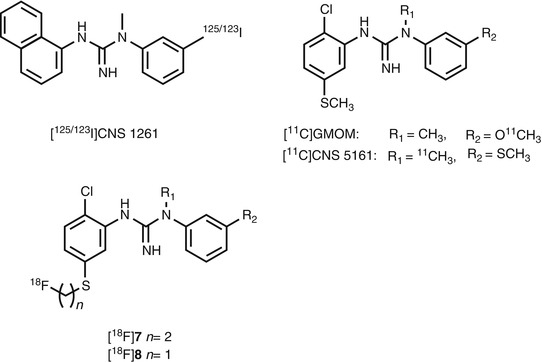
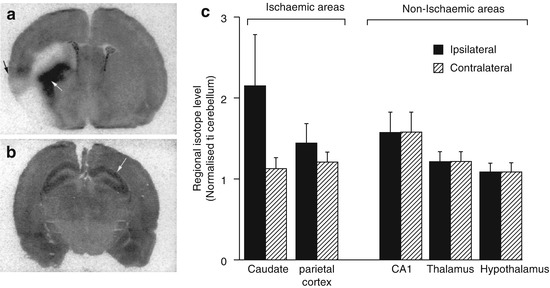
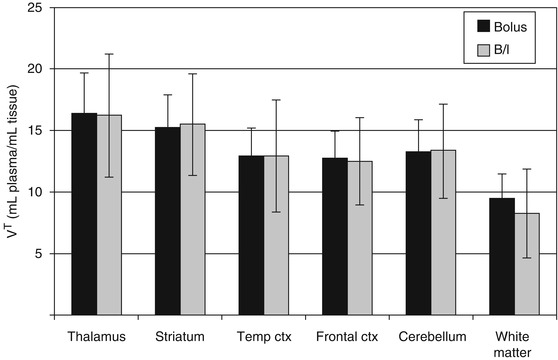
Fig. 18.10
Chemical structure of radiolabeled diarylguanidine derivatives
Fig. 18.11
Ex vivo autoradiography of [125I]CNS 1261 in coronal sections of rat brain at the level of (a) caudate nucleus (white arrow) and circumscribed zones of cerebral cortex (black arrow) (b) CA1 region of hippocampus (white arrow). The animals were injected with [125I]CNS 1261 15 min after permanent occlusion of the middle cerebral artery (left hemisphere) and sacrificed 120 min after tracer injection. (c) Accumulated radioactivity levels in ROIs to cerebellum (Owens et al. 2000)
Fig. 18.12
The mean V T values (±SD) of [125I]CNS 1261 for ROIs from healthy volunteers scanned with the bolus protocol (n = 7) (dark bars) and with the bolus/infusion protocol (n = 6) (light bars) (Bressan et al. 2004)
To provide potential PET radioligands for the PCP-binding site, [11C]-labeled diarylguanidine derivatives have been synthesized and evaluated. [11C]GMOM (Fig. 18.10) with a high affinity for NMDARs (K i = 5.2 nM) was prepared from the corresponding hydroxyl precursor with a specific activity of 46 GBq/μmol. In rats, brain uptake of [11C]GMOM was high and peaked during the early part of the study. Regional brain distribution at 10 min was 1.23, 0.93, and 0.74 % ID/g for the frontal cortex, hippocampus, and cerebellum, respectively, as regards the appearance of contrast in the NMDAR-rich and NMDAR-poor regions. Pretreatment with open channel blockers GMOM or MK-801 (1 mg/kg) or the NB2B-selective NMDAR modulator Ro 25–6981 led to a moderate and uniform decrease of [11C]GMOM uptake in brain regions. On the other hand, NMDAR co-agonist d-serine increased [11C]GMOM binding in all regions. PET imaging in baboons revealed that [11C]GMOM exhibited high BBB penetration. However, the brain distribution of [11C]GMOM was almost homogenous and pre-administration of MK801 did not reduce regional V T ’s or regional V T ratios. The authors hypothesized that these inconsistent results could be caused by the blocking effect of anesthesia (ketamine and isoflurane) to NMDAR activity (Waterhouse et al. 2004). Further PET investigations of [11C]GMOM in other conditions, such as in a conscious state, have not been reported so far.
Another diarylguanidine derivative, CNS 5161 (Fig. 18.10), has been shown to exhibit high affinity for the PCP-binding site (K i = 1.9 nM) (Hu et al. 1997; Dumont et al. 2002). Clinical trials using CNS 5161 have been evaluated in the treatment of neuropathic pain (Walters et al. 2002; Forst et al. 2007). The [3H]CNS 5161 showed a heterogeneous in vivo brain distribution in rats and a cortex/cerebellum ratio of 1.4. Pretreatment of NMDA increased the ratio in the hippocampus/cerebellum to 1.6–1.9, while MK801 diminished this increase that resulted in ratios close to 1. [3H]CNS 5161 appeared to be unstable in arterial blood, as 65 % of the parent compound was metabolized to several more hydrophilic compounds even 20 min after tracer injection (Biegon et al. 2007). The [11C]CNS 5161 was synthesized by [11C] methylation of the desmethyl guanidine precursor with low specific activity (41GBq/mmol) (Zhao et al. 2006). In human PET studies, [11C]CNS 5161 exhibited the largest uptake in the putamen and thalamus and the lowest uptake in the cerebellum, but relatively lower radioactivity was observed in the hippocampus compared with other gray matter regions. Similar to rats, the metabolism of [11C]CNS 5161 was rapid in human plasma. No blocking studies were reported in the literature (Asselin et al. 2004). Recently, clinical PET studies of [11C]CNS 5161 were carried out to assess the effect of levodopa on striatal and cortical NMDAR activity in patients with Parkinson’s disease with and without dyskinesias (Ahmed et al. 2011). The levodopa-treated dyskinetic patients had higher [11C]CNS 5161 uptake in the caudate, putamen, and precentral gyrus compared with patients without dyskinesias. These results are consistent with reports that upregulation of NMDARs has been associated with the development of levodopa-induced dyskinesias in animal models of Parkinsonism (Oh et al. 1998). However, no significant increase in [11C]CNS 5161 uptake was observed in patients with Parkinson’s disease when compared with control subjects. In addition, relatively high uptake of [11C]CNS 5161 in the cerebellum was observed, which was inconsistent with the brain distribution of [11C]CNS 5161 in a previous clinical PET study reported by Asselin et al. (2004). Further investigations are necessary in order to prove the usefulness of these diarylguanidines as radioligands for measuring NMDAR function by PET or SPECT.
Recently, 18F-labeled S-fluoroalkyl diarylguanidines were synthesized and used for in vitro evaluation. Among them, [18F]7 and [18F]8 showed high affinity for the PCP-binding site (K d = 2.4 nM for [18F]7 and K d = 1.4 nM for [18F]8). Further studies could not be performed on [18F]8 owing to low stability of this ligand. A broad receptor-binding assay for 60 receptors demonstrated that [18F]7 had high selectivity for the PCP-binding site of NMDARs (Robins et al. 2010). No in vivo studies of [18F]7 have been reported so far.
18.2.3 Other Open Channel Blocker Derivatives
The 6,11-ethanobenzo[b]quinolizinium derivatives are highly hydrophilic compounds reported to exhibit high affinity and specificity to the PCP-binding site of the NMDARs (Mallamo et al. 1994). Based on the SAR studies, new fluorine-18 radioligands ([18F]9 and [18F]10) and a carbon-11 ligand ([11C]11) have been developed as novel PET radioligands for the PCP-binding site of NMDARs as shown in Fig. 18.13 (Sasaki et al. 1998; Ishibashi et al. 2000). These compounds showed moderate binding affinity for the PCP-binding site (9: IC50 = 47 nM, 10: IC50 = 89 nM, 11: IC50 = 19 nM vs [3H]TCP) (Sasaki et al. 1998). In the preliminary radiosynthesis of [18F]9 and [18F]10, nucleophilic radiofluorination of mesylate was unsuccessful probably due to trapping of 18F− anion by the ammonium cation. An alternative synthetic route, including early fluorination of dienophile prior to the Diels-Alder reaction permitted successful radiosynthesis of [18F]9 and [18F]10 (Sasaki et al. 1998; Ishibashi et al. 2000). However, the specific activity of [18F]9 and [18F]10 was relatively low (>263 MBq/μmol). The [11C]11 was prepared by O-methylation of the hydroxyl precursor. The specific activity of [11C]11 was about 63 GBq/mmol. Values were approximately a 1,000-fold lower than the theoretical specific activity of C-11 ligands starting from [11C]CO2. In vivo biodistribution studies demonstrated that these three radioligands showed a quite low BBB permeability (0.02 − 0.05 % ID/g at 15 min) presumably because of their cationic property and extremely low lipophilicity (Ishibashi et al. 2000). To improve brain uptake and receptor specificity, the reduced derivative [11C]12 was developed as a prodrug for [11C]11. In the preliminary in vivo studies, cationic [14C]12 was easily metabolized to neutral [14C]11 at 15 min after injection. However, most of [14C]11 was cleared from the brain at 45 min. Although [11C]12 showed a higher BBB penetration (0.7 − 0.8 % ID/g at 15 min) than that of [14C]11, no regional difference was observed between the forebrain regions and cerebellum (Sasaki et al. 2001). Although [11C]12 was not confirmed to be a useful PET ligand for NMDARs, this prodrug strategy can be applied to the development of in vivo imaging agents.
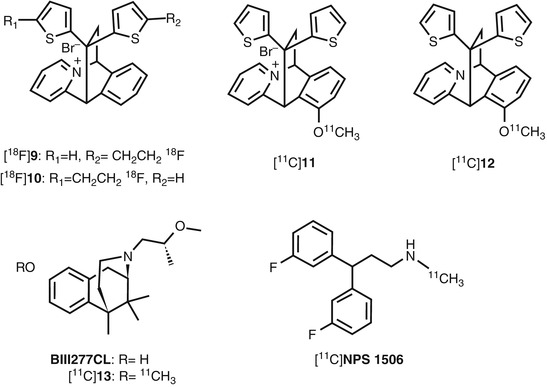
Fig. 18.13
Chemical structure of other radiolabeled open channel blocker derivatives
BIII277CL (Fig. 18.13), a potent anticonvulsant and neuroprotective agent, has been reported to have high affinity (K i = 4.5 nM vs [3H]MK-801) at the PCP-binding site of NMDARs (Grauert et al. 1997, 1998). Kokic et al. have developed a methyl analog of BIII277CL (13, Fig. 18.13) with substantial affinity for the PCP-binding site (K i = 49 nM vs [3H]TCP) as a candidate imaging agent for NMDARs. [11C]13 was prepared by O-[11C] methylation of BIII277CL with a specific activity of 30–60 GBq/μmol. PET studies of [11C]13 in pigs revealed homogenous localization in the cortex and cerebellum. The kinetic analysis using one-tissue compartment model revealed that V T of [11C]13 was approximately 1 in both the cortex and cerebellum. These results indicated a lack of specific binding of [11C]13 for NMDARs (Kokic et al. 2002).
NPS 1506 (Fig. 18.13) has been reported noncompetitive NMDAR antagonist with moderate affinity (IC50 = 664 nM vs [3H]MK-801). The NPS 1506 is neuroprotective in a variety of animal models of stroke and head injury with no PCP-like psychotomimetic effects at doses between 1 and 5 mg/kg i.p. in rats (Mueller et al. 1999). Considering such a pharmacological profile, which is different from the previous NMDAR antagonists, NPS 1506 was labeled with carbon-11 and evaluated as a novel PET tracer for the PCP-binding site of NMDARs. The [11C]NPS 1506 was prepared by N-[11C] methylation of primary amine precursor with a specific activity of around 50 GBq/μmol. Biodistribution of [11C]NPS 1506 in mice and rat demonstrated that uptake into the brain was rapid and occurred at high levels. However, the regional brain distribution was fairly uniform and did not clearly reflect the known localization of NMDARs in the rodent brain. In addition, treatment with unlabeled NPS 1506 did not cause any change in uptake in the mouse brain compared with the control group. An activator of NMDARs, 3-nitropropionic acid (20 mg/kg), did not produce any change in the regional uptake in the hippocampus nor the striatum. Therefore, [11C]NPS 1506 may be an unsuitable in vivo tracer for NMDARs because of its large nonspecific binding fraction and low in vitro binding affinity for NMDARs (Fuchigami et al. 2003).
18.3 Radioligands for Glycine Site
Because glycine and/or d-serine are essential co-agonists for NMDA ion-channel activation, various classes of glycine-binding site antagonists (Fig. 18.14) have been reported as anticonvulsant and neuroprotective agents (Danysz and Parsons 1998). Hypofunction of NMDARs is considered to be associated with the pathophysiology of schizophrenia. Indeed, it was reported that the enhancement of NMDAR activity via glycine site was efficacious in the treatment of schizophrenia (Millan 2005). Imaging of the glycine site of NMDARs was considered useful for obtaining information about the functional mechanism of the glycine site in the living brain and in the diagnosis of various neurological disorders.
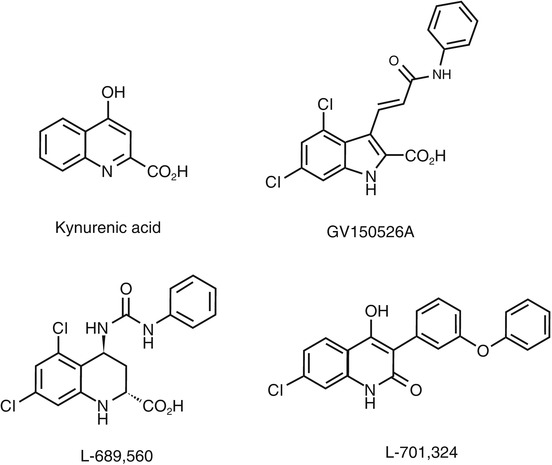
Fig. 18.14
Chemical structure of glycine-binding site antagonists
18.3.1 Radioligands Based on Cyclic Amino Acids
7-Chloro-5-iodokynurenic acid (14, Fig. 18.15) has been identified as a potent (IC50 vs [3H]glycine binding, 32 nM) and selective antagonist for the glycine site of NMDARs (Leeson et al. 1991). The [123I]14 was successfully synthesized by halogen exchange reaction of a mixture of 5-bromo-7-chloro- and 7-bromo-5-chlorokynurenic acid at a specific activity of 37 GBq/μmol. In the biodistribution studies in mice, [123I]14 showed fast deiodination (85 % of parent compound was changed to 123I−) and low brain uptake (0.61 % ID/g at 5 min). The [123I]14 had been demonstrated to be an unsuitable in vivo tracer (Dumont and Slegers 1997).
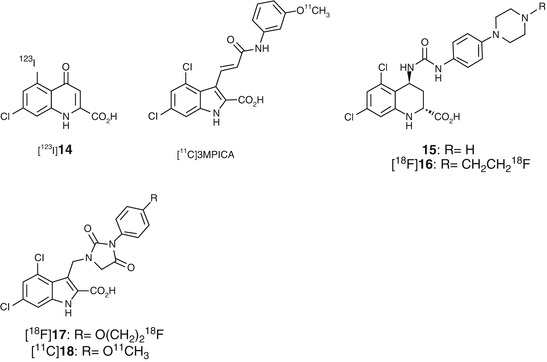
Fig. 18.15
Chemical structure of radiolabeled cyclic amino acid derivatives
GV150526A (Fig. 18.14) is one of the most potent glycine site antagonists (K i = 3.2 nM vs [3H]glycine) with reasonable lipophilicity (log P = 2.24) (Di Fabio et al. 1997). Based on the report, Waterhouse et al. developed a meta-methoxy derivative [11C]3MPICA (Fig. 18.15) with high affinity for the glycine site (K i = 4.8 nM vs [3H]MDL 105,519). The [11C]3MPICA was prepared by O-[11C] methylation of the corresponding desmethyl derivative with a specific activity of 44 GBq/μmol (Waterhouse et al. 2002a). In the biodistribution studies in rats, regional uptake of [11C]3MPICA at 2 min ranged from 0.11 to 0.18 % ID/g. The radioactivity cleared rapidly from all brain regions examined. Blocking studies using unlabeled compound (1 mg/kg) did not cause a marked reduction in the ratio of the brain tissues/blood. Treatment with warfarin (100 mg/kg) led to reduction in blood radioactivity concentrations while brain uptake was not changed significantly, which indicated that factors other than binding to serum albumin could prevent BBB penetration of [11C]3MPICA (Waterhouse et al. 2002b).
Trans-5,7-dichloro-4-substituted-2-carboxytetrahydroquinolines such as 15 (Fig. 18.15) showed high affinities (K i = 6.0 nM vs [3H]5,7-dichlorokynurenic acid) for the glycine site of NMDARs (Leeson et al. 1992). Based on the SAR studies, fluoroethyl derivative [18F]16 (Fig. 18.15) was designed, synthesized, and evaluated as a PET radioligand for glycine site. The 16 had a relatively low lipophilicity (logD = 1.3) and high affinity (K i = 12 nM vs [3H]MDL 105,519) for the glycine-binding site. The radiosynthesis of [18F]16 was achieved by N-[18F]fluoroethylation of a piperidine precursor with 2-[18F]fluoroethyltosylate followed by hydrolysis of methyl ester. Biodistribution studies revealed that [18F]16 showed homogenous low levels of accumulation in all considered brain regions. Because the difference in regional brain distribution of [18F]16 could not be detected, no blocking experiments were performed (Piel et al. 2003). Recently, hydantoin-substituted indole carboxylic acids were developed as novel PET ligands for the glycine-binding site with a higher lipophilicity than 16. Compound 17 and 18 showed moderate affinity for the glycine site (K i = 53 nM for 17 and K i = 31 for 18 in [3H]MDL 105,519 binding assay). The logD values of 17 and 18 were 2.07 and 2.26, respectively. [18F]17 and [11C]18 were successfully synthesized with overall decay-corrected radiochemical yields of 5–7 and 6–9 % and specific activities of 24–67 GBq/μmol and 8–26 GBq/μmol, respectively (Bauman et al. 2011). No further in vivo evaluations of these radioligands have been reported so far.
18.3.2 5-Aminomethylquinoxaline-2,3-Dione Derivative
(D)-7-Bromo-N-(1-phosphonoethyl)-5-aminomethylquinoxaline-2,3-dione (PAMQX, Fig. 18.16) has been discovered as a selective and high-affinity glycine site antagonist with an IC50 value of 6 nM against [3H]MDL-105519 binding (Auberson et al. 1999). The 7-iodo derivative of PAMQX (19, Fig. 18.16) exhibited comparable affinity (IC50 = 8 nM vs [3H]MDL-105,519) for the glycine site with PAMQX (Ametamey et al. 2000). Radiosynthesis of [131I]19 was achieved by iododestannylation of N-CBZ-protected tributylstannyl precursor followed by deprotection of CBZ group. Biodistribution studies of [123I]19 in mice revealed an extremely low BBB permeability of [123I]19 (0.03 % ID/g at 30 min) probably due to its high polarity and low log D value (logD 7.4 = −2.5) (Ametamey et al. 2000).
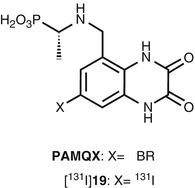
Fig. 18.16
Chemical structure of PAMQX and radioiodinated PAMQX derivative
18.3.3 4-Hydroxyquinolone Derivatives
The radioligands consisting of carboxylic acids showed poor brain penetration due to the highly polar nature of the charged carboxylate. The 4-hydroxyquinolones (4-HQs)-devised carboxylic bioisostere had been developed as one of the most potent antagonists for the glycine-binding site of NMDARs. As expected, this series showed in vivo anticonvulsant activity, which suggested an ability to penetrate the BBB (Kulagowski et al. 1994; Rowley et al. 1997). In line with this, Morris et al. synthesized [11C]L-703,717 (Fig. 18.17), one of the most potent glycine site antagonists (IC50 = 4.5 nM vs [3H]L-689,560), by O-[11C] methylation of phenol precursor using [11C]CH3I containing K2CO3/Kryptofix 2.2.2 as a base, but the radiochemical yield was very low (7 %) (Morris et al. 1997). The labeling precursor has three possible sites (amide, enol, and phenol) which could be reacted with [11C]CH3I. Therefore, regioselective methylation at the phenol position was required to prepare [11C]L-703,717 for high yield and purity. Haradahira et al. succeeded in the synthesis of [11C]L-703,717 by using 5 eq of NaH as a base in high yields (87 %) with a specific activity of 47–53 GBq/μmol. The excess amount of NaH can give the triply deprotonated species, then the most accessible terminal phenol can be preferentially methylated (Haradahira et al. 1999). In the biodistribution study in mice, [11C]L-703,717 showed low BBB permeability (0.32 − 0.36 % ID/g at 1 min) and extremely high blood concentration presumably due to high affinity for the warfarin-binding site of plasma protein. Indeed, warfarin led to a dose-dependent enhancement of the initial brain uptake of [11C]L-703,717 that resulted in a fivefold increase in the brain radioactivity at the dose of 200 mg of warfarin. However, the radioactivity in the cerebrum was rapidly decreased (0.2 % ID/g) with some retention observed in the cerebellum (0.65 % ID/g) at 30 min after injection. This suggested inconsistent binding with known distribution of NMDARs. In addition, a significant blocking effect by nonradioactive L-703,717 (2 mg/kg) was observed only in the cerebellum (Haradahira et al. 2000). To improve brain penetration of [11C] L-703,717, acetyl derivative of L-703,717 ([11C]20, Fig. 18.17) was prepared and evaluated as prodrug of [11C]L-703,717. Initial brain uptake of [11C]20 (0.71 % ID/g) was twofold higher than that of [11C]L-703,717 (0.36 %ID/g). Metabolism experiments of [11C]20 in rat brain homogenates confirmed that about 80 % of [11C]20 was converted to [11C]L-703,717 at 20 min after injection. Ex vivo autoradiography demonstrated that [11C]20 exhibited similar cerebellar localization of radioactivity to [11C]20 (Haradahira et al. 2001). Further in vivo evaluation revealed that the specific cerebellar binding of [11C]L-703,717 was abolished in the NR2C-deficient mice as shown in Fig. 18.18a–c. This result suggested that the in vivo cerebellar localization of [11C]L-703,717 could be caused by preferential binding to the NR2C subunit-containing NMDARs predominantly expressed in the cerebellum. However, [11C]L-703,717 exhibited high specific localization in the hippocampus and cerebral cortex under in vitro condition as shown in Fig. 18.18d–f, indicative of the absence of intrinsic selectivity for the NR2C subunit-containing NMDARs in the cerebellum (Haradahira et al. 2002b). Thus, the authors next hypothesized that endogenous NMDAR co-agonists might play a key role in the unusual localization in the cerebellum of [11C]L-703,717. Both glycine and d-serine are known as co-agonists of NMDARs and exist in micromolar level. Glycine is distributed throughout the brain, while d-serine is localized only in the forebrain regions (Hashimoto et al. 1993; Schell et al. 1997). d-serine is reported to be nearly undetectable in cerebellum due to the highest level of d-amino acid oxidase (DAO) activity (Schell et al. 1995). Haradahira et al. demonstrated that the cerebellar localization of [11C]L-703,717 was abolished in mutant ddY/DAO– mice, in which the d-serine level in cerebellum was markedly increased (Fig. 18.19). Hence, the authors proposed that the low level of [11C]L-703,717 binding in forebrain regions could be explained by the strong inhibition of high amount of endogenous d-serine; whereas, specific localization in the cerebellum might be due to the lack of d-serine (Haradahira et al. 2003). Considering that [11C]L-703,717 has been demonstrated to recognize cerebellar NMDARs to some extent, clinical PET studies of [11C]20 (a prodrug of [11C] L-703,717) were performed for the visualization of NMDARs in the cerebellum (Matsumoto et al. 2007). Binding potential (BP) calculated from white matter as a reference region in the cerebellar cortex was twofold higher than in the cerebral cortices (cerebellar cortex; BP = 2.2, cerebral cortices; BP = 1.1). Regional brain distributions were consistent with previous reports of rodents (Haradahira et al. 2001). However, the brain uptake of [11C]20 was too low for the visualization of the cerebellar NMDAs by PET (Matsumoto et al. 2007).
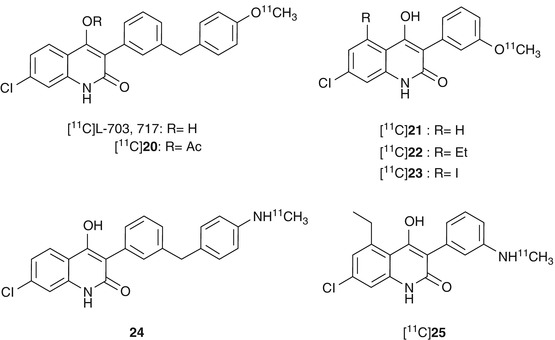
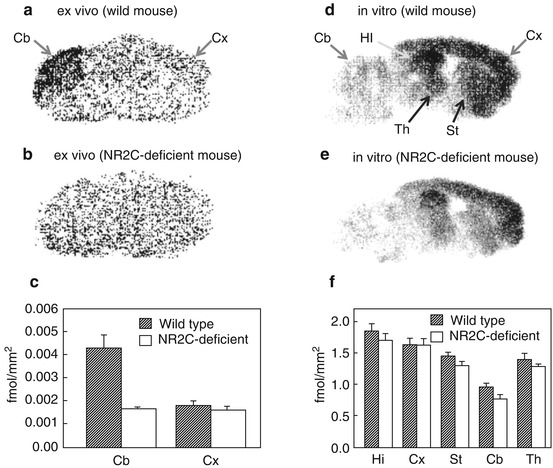
Fig. 18.17
Chemical structure of radiolabeled 4-hydroxyquinolone derivatives
Fig. 18.18
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Tracers for Beta-Amyloid and Other Proteinopathies
 PET Imaging of ABC Transporters in the BBB
PET Imaging of ABC Transporters in the BBB
 Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT




Ex vivo and in vitro autoradiograms of [11C]L-703,717 in sagittal sections of wild-type and NR2C-deficient mice brain. (a, b) Ex vivo images obtained at 20 min after injection of [11C]L-703,717 and warfarin (60 mg/kg). (c) Quantified values of the ex vivo binding in cerebral cortex (Cx) and cerebellum (Cb). (d, e) In vitro images of [11C]L-703,717. (f) Quantified values of the in vitro bindings in hippocampus (Hi), cerebral cortex (Cx), striatum (St), cerebellum (Cb), and thalamus (Th) (Haradahira et al. 2002b)
Related posts:
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Imaging of ABC Transporters in the BBB
Imaging Type 1 Glycine Transporters in the CNS Using Positron Emission Tomography
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
