DEVELOPMENTAL CRANIOFACIAL ANOMALIES: SYNDROMIC, CLEFTS, ENCEPHALOCELES (BRAIN HETEROTOPIAS), AND NASAL DERMOIDS
KEY POINTS
- Computed tomography and magnetic resonance imaging are critical to medical decision making in developmental abnormalities of this region and can help anticipate syndromic relationships of this as well as other organ systems.
- Reports must be logically constructed and comprehensive, anticipating all anatomic and pathologic that might lead to altered treatment plans.
Craniofacial syndromic dysmorphic changes are often associated with functional disabilities of the associated anatomy devoted to the special senses or those related to other systems such as the skin and central nervous system. The dysmorphic features must be recognized clinically. A minority of the patients are then brought to imaging as part of a plan to identify possibly associated deranged anatomy that cannot be directly observed but may predict or explain functional deficits. Imaging may also be used as a tool to plan cosmetic improvements.
Clues to the presence of a syndrome include cervical and facial dysmorphism, visual system anomalies, endocrine dysfunction, cardiac problems, renal abnormalities, and hair and skin changes.1 Genomic anomalies have been established for many syndromic and nonsyndromic disorders.
Genetic associations may help to anticipate and limit the damage of possibly associated genetic errors and, in the future, perhaps lead to some preventive strategies. Such information already facilitates genetic counseling. Recognizing the common pathways of genomic errors can also lead to prompt evaluation of other organ systems dependent for normal development on that part of the genetic code and perhaps limit morbidity due to those errors. For instance, since ear malformations are associated with an increased frequency of renal anomalies, renal ultrasound is often obtained if a syndromic etiology is suspected. Such recognition may also yield a diagnosis that had not previously been suspected. Examples include cochlear anomalies seen as characteristic of the branchiootorenal (BOR) syndrome and the combination of ocular, nasal, and inner ear findings of the CHARGE syndrome.
Isolated cleft palate is a very common occurrence that occurs in approximately 1 in 2,000 live births.2 It is only syndromic in 50% of cases.2 The tasks for the diagnostic imager are limited in this condition in which diagnosis is straightforward and repairs are fairly standardized.
Developmental encephaloceles and ectopic brain tissue are largely isolated events but may accompany other defects. Their clinical presentation varies with age. Almost all will require repair.
Nasal-region dermoids and their related tracts and draining sinuses are primarily a pediatric age-range problem presenting a fairly discreet range of appearances and potential problems. These tend to not be syndromic, but they may be accompanied by other developmental problems.
GENERAL EMBRYOLOGY
The various degrees of facial dysplasia and dysgenesis observed with imaging are growth errors and arrests of very complex interrelated systems. The following summary should explain, at least globally, the interrelated nature of the development of the face, skull, brain, and special senses systems.
The development of skull combines that of the neurocranium including its membranous and cartilaginous components; visceral cranium that includes the maxilla, palate, and mandible; and development of sutures and related synchondroses.
The development of the face depends on five facial prominences. Neurogenic placodes form the olfactory, optic (Chapter 46), and otic (Chapter 107) structures. The pharyngeal arches and pouches form the related structures of the pharynx. All of this morphogenesis is under a high level of molecular regulation. This regulation modulates the behavior of genes that control systems such as the facial rhombomeres and neural crest elements. Errors in some of these systems result in specific known syndromes such as the neurocristopathies that include Treacher Collins syndrome (mandibulofacial dysostosis), Pierre Robin and DiGeorge sequences, and hemifacial microsomia. These factors interact with molecular regulation of skeletal morphogenesis such as fibroblast growth factors and receptors. The timing of the specific errors in these sequences of events will influence the degree of expressed morphologic errors and possible associations.
The migratory pathways of various germ cell layers cross each other during this extraordinarily complex process and are sometimes left or kept open along the way. This creates the opportunity for the formation of dermoid cysts and their related tracts, meningoencephaloceles and the less common occurrence of abnormal brain tissue, endodermal remnants such as the Rathke cleft, or normal tissue such as the anterior pituitary lobe being left where it does not belong.
The first 12 weeks of fetal growth are the most critical period in the embryology of the face. Neural tube development and closure is between the third and fourth weeks of fetal growth. The neural crest cells migrate laterally and anteriorly around the eye to the frontonasal process as the neural tube is formed. These neural crest cells in the face primarily form the mesenchymal cells that provide the bone, cartilage, and muscles of the face.
Transient spaces exist between the neural crest aggregates that form the skull base and nose. These spaces become the conduits for developmental midline nasal masses. These spaces are named the fonticulus frontalis, the prenasal space, and the foramen cecum. The fonticulus nasofrontalis lies between the frontal and nasal bones. The prenasal space lies between the nasal bones—the precursor of the septum and nasal cartilages. These spaces typically fuse and ossify. When development is abnormal, dermoids, ectopic brain tissue, and encephaloceles result. Similar important conduits in the central skull base include the intrasphenoidal synchondrosis and the craniopharyngeal canal.
ANATOMY: COMPUTED TOMOGRAPHY APPEARANCE OF THE DEVELOPING ANTERIOR SKULL BASE FROM BIRTH TO 2 YEARS OF AGE
The anterior skull base at birth is made up of the not yet ossified cartilage of the perpendicular plate of the ethmoid bone and crista galli, an essentially largely not ossified roof of the nasal cavity, and partially ossified ethmoidal bone.3,4 The majority of the cartilage of the anterior skull base will be ossified by 2 years of life (Fig. 79.1). Understanding this period of the normal anterior skull base development is important to avoid misinterpretating normal anatomic variations for a pathologic condition. Such understanding also highlights the limitations of computed tomography (CT) for evaluating the anterior skull base in infants and toddlers.
The ethmoidal labyrinth and turbinates are the nidus for ossification of the anterior skull base. Ossification proceeds from this area toward the midline over the first few months of life, resulting in the midline gaps normally seen on coronal CT scans in this region. The ossification of the anterior skull base will proceed relatively consistently with fusion of the cribriform plate and lateral ethmoid bone masses beginning as early as 2 months of age. Ossification of the paired ethmoidal labyrinths spreads to the crista galli and perpendicular plate.4
ANATOMY: COMPUTED TOMOGRAPHY APPEARANCE OF THE DEVELOPING CENTRAL SKULL BASE
The spheno-occipital synchondrosis base allows for growth of the skull base up to the time of skeletal maturity, typically in the mid teenage years (Fig. 79.2A). It is of only limited interest in congenital malformations.
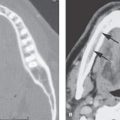
The intrasphenoidal synchondrosis is in the midbody of the sphenoid bone just below the planum sphenoidale (Fig. 79.2A). It is opened at birth but closes to a sclerotic remnant that is barely visible by about 2 years of age. This can be a pathway of a central meningoencephalocele or site of ectopic brain tissue (Figs. 8.23A,B and 79.3).
The craniopharyngeal canal extends from the roof of the nasopharynx to the floor of the sella during development (Fig. 79.2A). It is the pathway of migration for the endoderm Rathke cleft; therefore, the anterior pituitary gland remnants of both structures may be left anywhere along this migratory pathway (Fig. 79.2A–D). The tubular channel may persist indefinitely but is generally obliterated by bone that can be seen as a round sclerotic focus.

FIGURE 79.1. Coronal computed tomography images from newborn through 2 years of age to demonstrate mineralization of the anterior skull base. A: The cribriform plate and ethmoid roof are not mineralized at their junction with the frontal bone (F). There is mineralization of the ethmoid bones medial walls just beginning (arrow). B: At about 1 year of age, there is mineralization of the ethmoid roof and early appearance of mineralization of the crista galli (arrows). C: At approximately 2 years of age, the anterior skull base is normally very close to fully mineralized. (From Belden CJ, Mancuso AA, Kotzur IM. The developing anterior skull base: CT appearance from birth to 2 years of age. AJNR Am J Neuroradiol. 1997;18(5): 811–818, with permission.)
SPECIFIC GENETIC SYNDROMES AND REPORTING
Specific Conditions
The following list serves as a general, although incomplete, reference to a basic classification of these syndromes and whether or not they occur with enough frequency to be included in a resource such as this and, if so, where they may be found in this resource.
- Those with primarily ocular consequences are discussed in Chapter 46. These include Norrie disease, Walker-Warburg, trisomy 13, and CHARGE.
- Those with associated hearing deficits are discussed in Chapter 107. These include the oculoauriculovertebral (OAV) spectrum that encompasses hemifacial microsomia, Goldenhar syndrome, the first and second branchial arch syndrome, and facioauriculovertebral syndrome. This group also includes the otocraniofacial or craniosynostosis syndromes such as Crouzon disease or otocraniofacial dysostosis and Apert syndrome acrocephalosyndactyly. The Robin sequence, BOR syndrome, and Treacher Collins syndrome are also included in Chapter 107.
- A discussion of craniosynostosis in general terms is beyond the scope of this resource. Recall that it can be syndromic or nonsyndromic, with the syndromic variety causing much greater treatment challenges.
- Cleft palate can be both syndromic or nonsyndromic, so it is described with other cleft and facial anomalies that are more sporadic in nature in Chapters 80 and 81.
- Facial anomalies associated with primary problems of the integumentary system are not discussed in this resource.
- Anomalous development related to the phacomatoses is discussed along with masses and/or vascular anomalies seen in conditions such as neurofibromatosis type 1 and Sturge-Weber syndrome.
- Syndromes expressed as bony dysplasias and/or with bony masses are discussed along with facial skeletal abnormalities.

FIGURE 79.2. Two patients presenting with respiratory obstruction in the newborn period. A, B: Patient 1. A magnetic resonance imaging (MRI) study was done when a mass was seen at endoscopy. In (A), the T2-weighted (T2W) sagittal image shows a cyst that obstructs the posterior nasal cavity and nasopharynx and contacts the central skull base at the outlet (black arrowhead) of the craniopharyngeal canal (white arrowhead). That canal should be distinguished both from the intrasphenoidal synchondrosis (white arrow) and the spheno-occipital synchondrosis (black arrow). In (B), the T2W coronal image shows the cyst to extend into the opening of the craniopharyngeal canal (arrow). C, D: Patient 2 presenting with noisy respiratory efforts. MRI was done because of a submucosal mass observed in the nasopharynx at endoscopy. In (C), the T1-weighted (T1W) sagittal image shows a somewhat dysmorphic pituitary gland with no good differentiation between the anterior and posterior lobes (arrow). In addition, the craniopharyngeal canal is widened and leads to abnormal tissue within the nasopharynx occluding the airway above the soft palate (arrowheads). In (D), the T1W coronal image shows the pituitary gland in a relatively normal position but dysmorphic abnormal tissue along the craniopharyngeal canal and projecting into the nasopharynx (arrowheads).
Related posts:
 Preseptal Compartment: Acute and Chronic Infections and Noninfectious Inflammatory Conditions
Preseptal Compartment: Acute and Chronic Infections and Noninfectious Inflammatory Conditions
 Necrotizing (“Malignant”) Otitis Externa
Necrotizing (“Malignant”) Otitis Externa
 Hypopharynx: Developmental Abnormalities
Hypopharynx: Developmental Abnormalities
 Tumors of the Submandibular Gland and Space and Tumorlike Conditions
Tumors of the Submandibular Gland and Space and Tumorlike Conditions
 Masticator Space, Buccal Space, and Infratemporal Fossa Infections and Other Inflammatory Conditions
Masticator Space, Buccal Space, and Infratemporal Fossa Infections and Other Inflammatory Conditions
 Oral Cavity and Floor of the Mouth: Malignant Tumors
Oral Cavity and Floor of the Mouth: Malignant Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


