Spinal arteriovenous shunts (SAVSs) are rarely diagnosed in infants and children, but they are important clinically because morbidity can be significant. Although these lesions do not form a distinct pathologic group separate from the SAVSs seen in older patients, experience with these malformations in the pediatric population has led to the identification of several important features that are characteristic of this group of SAVSs. Association with genetic abnormalities, heritable (hereditary hemorrhagic telangiectasia) and nonheritable somatic (spinal arteriovenous metameric syndrome or Cobb syndrome), is relatively common and likely underrecognized. Male predominance is more pronounced than in the adult population. Hemorrhagic presentation is more frequent than in adults, except in extremely young children. The natural history seems to be better than previously thought, with early rehemorrhage uncommon. Despite early presentation and severe symptoms, these lesions are frequently amenable to endovascular treatment, often with anatomic cure achieved and with improvement or stabilization of symptoms after partial targeted treatment.
Spinal arteriovenous shunts (SAVSs) are distinctly uncommon lesions in all age groups, but presentation in infants and children is rarer still. When SAVSs are diagnosed in pediatric patients, they are frequently associated with significant morbidity and their treatment is often challenging. The SAVSs seen in infants and children are not generally unusual or unique lesions from a morphologic standpoint, however. Early presentation of a SAVS in an infant or child may thus represent a particularly severe manifestation of disease processes that more commonly present in older patients. In other words, the SAVSs seen in the pediatric population may represent a more complete phenotypic expression of defects affecting the spinal circulation that manifest more frequently in a delayed and isolated manner. Seen in this light, the topic of this review—an analysis of the features that lead to early presentation of a SAVS in an infant or child less than 16 years of age—may shed light not only on SAVSs in the pediatric population but on all SAVSs, including those presenting in adults.
Incidence of pediatric spinal arteriovenous shunts
The true incidence of SAVSs is not known. It has been estimated that SAVSs comprise 10% of all central nervous system (CNS) arteriovenous shunts (AVSs) in patients of all ages . The incidence of brain arteriovenous malformations (BAVMs) in North America is 1.34 per 100,000 persons, and SAVSs are identified annually in approximately 1 in 1 million patients . Diagnosis of a SAVS in a child is even less frequent. In the senior author’s series of SAVSs seen at the Bicêtre Hospital, an international referral center for pediatric neurovascular disease in Paris, France, children accounted for 5% of all patients with CNS AVSs . Pediatric SAVSs comprised 20% of SAVSs in all ages seen by this same group . This number varies significantly from institution to institution relative to referral patterns and, of course, does not necessarily reflect incidence in the population as a whole. Whatever the true incidence of these lesions, it is likely that SAVSs are generally underdiagnosed because of their variable presentation and the complexity of diagnosis.
Classification of pediatric spinal arteriovenous shunts
The spinal cord and its circulatory system are significantly older phylogenetically than the cerebral circulation. The basic plan of spinal circulation—a fused ventral longitudinal channel supplemented by a perimedullary pial network supplied by segmental aortic branches—has remained relatively stable and unchanged throughout the ontogeny of vertebrates and across species. Profound genetic, molecular, or signaling errors that disrupt the normal developmental pathways, vasculogenesis, and vessel assembly in the spine during embryogenesis are likely most often lethal (here, the extreme eloquence of spinal cord tissue relative to volume likely also plays a role), and thus do not achieve high penetrance in the population. This may be one reason for the relative rarity of anatomic variation in the spinal (as compared with the cerebral) vasculature and possibly provides one explanation why SAVSs occur infrequently there. Certainly, the incidence of aneurysms is dramatically lower in the spine compared with the cerebral circulation . Whatever the reasons for their rarity, it is likely SAVSs are the result of a range of underlying biologic defects that result in a susceptibility to form a pathologic condition, such as an AVS, but that require one or two triggering events after embryogenesis to achieve complete phenotypic expression, as has been proposed for other CNS vascular lesions . In fact, there is evidence that a biologic defect is responsible for a large number of SAVSs, particularly those that present in the pediatric population. A recent series of SAVSs presenting in children younger than 2 years of age, for example, found that nearly two thirds of patients (62%) had evidence of a heritable genetic defect (hereditary hemorrhagic telangiectasia [HHT]) or a somatic nonheritable mutation (spinal arteriovenous metameric syndrome [SAMS]) ( Fig. 1 ) .
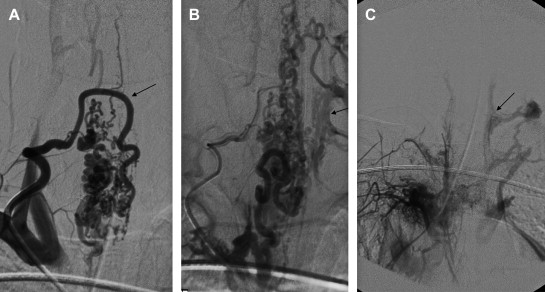
Several classifications have been used since SAVSs were first recognized as a clinical entity in the late 19th century . The classifications have been used inconsistently, and the terminology, which is primarily descriptive, is often confusing. The situation is similar to the inconsistent and imprecise descriptive nomenclature used previously to describe vascular birthmarks and vascular anomalies. In 1982, Mulliken and Glowacki focused on the endothelial cell characteristics of malformations and hemangiomas, clarified terminology, and proposed a classification that was grounded in the vascular biology of these lesions. Likewise, any classification of SAVSs, if it is to lead to better comprehension of these lesions, help to predict their natural history, and guide their treatment, must attempt to incorporate the vascular biology of these diseases.
The classification of SAVSs used here is the result of a 25-year single-center experience by the senior author treating SAVSs at the Bicêtre Hospital and has been outlined in numerous articles and book chapters ( Box 1 ) . First, a biologic basis for the SAVS is established and characterized. This may be a heritable mutation, such as one of the genetic defects associated with HHT, or a presumed somatic mutation if multiple lesions are present in a pattern consistent with SAMS . Klippel-Trenaunay and Parkes-Weber syndromes have also been associated with SAVSs, as have neurofibromatosis and Ehlers-Danlos syndrome type IV, among others . In the latter, however, the arterial wall disease focuses the lesion on the mural rupture of an artery into a venous plexus rather than a postcapillary vessel disorder. If no known genetic defect is present and the SAVS is not related to trauma, it is assumed to be an isolated sporadic SAVS. It is likely some of these isolated and apparently sporadic SAVSs may represent incomplete phenotypic expression of an underlying but undiagnosed genetic or segmental syndrome. Next, the morphology of the shunt is analyzed. A SAVS may be a fistula, consisting of a direct connection between the artery and vein, or a nidus, in which an intervening network of vessels is present. Fistulae are further subdivided into microfistulae and macrofistulae. The term angioarchitecture describes not only the morphology of a SAVS at a given time but places the lesion in a temporal sequence, recognizing that most SAVSs induce changes over time; marked venous ectasias may develop, and additional arterial pedicles may be recruited. The type of shunt, nidus, or fistula, remains fixed; however, with time, some fistulae may resemble nidal type malformations because of the extensive pial reflux or intense intrinsic network congestion. Finally, the location of the SAVS is identified: intradural, epidural, or parachordal/paraspinal. Epidural lesions frequently include bone and may be malformations. So-called “spinal dural” AVSs are acquired and are excluded from this review because they relate to a different adult disease entity . Intradural lesions may be superficial or deep in the cord. The type of shunt and its location have implications for the clinical presentation, treatment, and likelihood of complete cure with endovascular methods.
- A.
According to biologic abnormality
- 1.
Heritable genetic defect
HHT type 1 (endoglin [ENG]) 9q33-34
HHT type 2 (activin receptor-like kinase [ALK1]) 12q11-14
HHT type 3 (Smad 4) 5q31-32
- 2.
Nonheritable defect (somatic genetic) often segmental
SAMS
Klippel-Trenaunay syndrome with limb venolymphatic-associated lesions
Parkes-Weber syndrome
- 3.
Sporadic isolated arteriovenous lesion (forme fruste of 1 and 2)
- 4.
Acquired lesion (?)
- 1.
- B.
According to morphology of AVS
Nidus (spinal cord arteriovenous malformation [SCAVM])
Fistula (spinal cord arteriovenous formation [SCAVF])
Macrofistula
Microfistula
Multifocal intradural lesions
- C.
According to location of SAVS
Intradural
Intramedullary
Extramedullary
Radicular
Filum
Epidural AVS (intraspinal)
Parachordal arteriovenous formation (AVF)
Branchial AVF
Vertebrovertebral arteriovenous formation (VVAVF)
Paraspinal arteriovenous formation (PSAVF)
The pathophysiology of spinal cord arteriovenous shunts (SCAVSs) is poorly understood. Like BAVMs, they are not diagnosed antenatally, although they are congenital . Most likely, a patient is born with a propensity to form an AVS, which then appears after a second or third event interferes with vessel repair or angiogenesis, although this process remains to be clarified. Presumably, etiologic factors important in the formation of BAVMs are also present in SAVSs. BAVMs are thought to represent populations of structurally unstable blood vessels with phenotypic evidence of upregulated angiogenesis . Animal models suggest that errors in artery-vein identity, as dictated by the Notch signaling pathway, may also play a role . Investigators have filled in some of the gaps in our knowledge of the pathophysiology of SAVSs with work done on the genetics, vascular biology, and endothelial cell biology of HHT in human beings and animal models.
Classification of pediatric spinal arteriovenous shunts
The spinal cord and its circulatory system are significantly older phylogenetically than the cerebral circulation. The basic plan of spinal circulation—a fused ventral longitudinal channel supplemented by a perimedullary pial network supplied by segmental aortic branches—has remained relatively stable and unchanged throughout the ontogeny of vertebrates and across species. Profound genetic, molecular, or signaling errors that disrupt the normal developmental pathways, vasculogenesis, and vessel assembly in the spine during embryogenesis are likely most often lethal (here, the extreme eloquence of spinal cord tissue relative to volume likely also plays a role), and thus do not achieve high penetrance in the population. This may be one reason for the relative rarity of anatomic variation in the spinal (as compared with the cerebral) vasculature and possibly provides one explanation why SAVSs occur infrequently there. Certainly, the incidence of aneurysms is dramatically lower in the spine compared with the cerebral circulation . Whatever the reasons for their rarity, it is likely SAVSs are the result of a range of underlying biologic defects that result in a susceptibility to form a pathologic condition, such as an AVS, but that require one or two triggering events after embryogenesis to achieve complete phenotypic expression, as has been proposed for other CNS vascular lesions . In fact, there is evidence that a biologic defect is responsible for a large number of SAVSs, particularly those that present in the pediatric population. A recent series of SAVSs presenting in children younger than 2 years of age, for example, found that nearly two thirds of patients (62%) had evidence of a heritable genetic defect (hereditary hemorrhagic telangiectasia [HHT]) or a somatic nonheritable mutation (spinal arteriovenous metameric syndrome [SAMS]) ( Fig. 1 ) .
Several classifications have been used since SAVSs were first recognized as a clinical entity in the late 19th century . The classifications have been used inconsistently, and the terminology, which is primarily descriptive, is often confusing. The situation is similar to the inconsistent and imprecise descriptive nomenclature used previously to describe vascular birthmarks and vascular anomalies. In 1982, Mulliken and Glowacki focused on the endothelial cell characteristics of malformations and hemangiomas, clarified terminology, and proposed a classification that was grounded in the vascular biology of these lesions. Likewise, any classification of SAVSs, if it is to lead to better comprehension of these lesions, help to predict their natural history, and guide their treatment, must attempt to incorporate the vascular biology of these diseases.
The classification of SAVSs used here is the result of a 25-year single-center experience by the senior author treating SAVSs at the Bicêtre Hospital and has been outlined in numerous articles and book chapters ( Box 1 ) . First, a biologic basis for the SAVS is established and characterized. This may be a heritable mutation, such as one of the genetic defects associated with HHT, or a presumed somatic mutation if multiple lesions are present in a pattern consistent with SAMS . Klippel-Trenaunay and Parkes-Weber syndromes have also been associated with SAVSs, as have neurofibromatosis and Ehlers-Danlos syndrome type IV, among others . In the latter, however, the arterial wall disease focuses the lesion on the mural rupture of an artery into a venous plexus rather than a postcapillary vessel disorder. If no known genetic defect is present and the SAVS is not related to trauma, it is assumed to be an isolated sporadic SAVS. It is likely some of these isolated and apparently sporadic SAVSs may represent incomplete phenotypic expression of an underlying but undiagnosed genetic or segmental syndrome. Next, the morphology of the shunt is analyzed. A SAVS may be a fistula, consisting of a direct connection between the artery and vein, or a nidus, in which an intervening network of vessels is present. Fistulae are further subdivided into microfistulae and macrofistulae. The term angioarchitecture describes not only the morphology of a SAVS at a given time but places the lesion in a temporal sequence, recognizing that most SAVSs induce changes over time; marked venous ectasias may develop, and additional arterial pedicles may be recruited. The type of shunt, nidus, or fistula, remains fixed; however, with time, some fistulae may resemble nidal type malformations because of the extensive pial reflux or intense intrinsic network congestion. Finally, the location of the SAVS is identified: intradural, epidural, or parachordal/paraspinal. Epidural lesions frequently include bone and may be malformations. So-called “spinal dural” AVSs are acquired and are excluded from this review because they relate to a different adult disease entity . Intradural lesions may be superficial or deep in the cord. The type of shunt and its location have implications for the clinical presentation, treatment, and likelihood of complete cure with endovascular methods.
- A.
According to biologic abnormality
- 1.
Heritable genetic defect
HHT type 1 (endoglin [ENG]) 9q33-34
HHT type 2 (activin receptor-like kinase [ALK1]) 12q11-14
HHT type 3 (Smad 4) 5q31-32
- 2.
Nonheritable defect (somatic genetic) often segmental
SAMS
Klippel-Trenaunay syndrome with limb venolymphatic-associated lesions
Parkes-Weber syndrome
- 3.
Sporadic isolated arteriovenous lesion (forme fruste of 1 and 2)
- 4.
Acquired lesion (?)
- 1.
- B.
According to morphology of AVS
Nidus (spinal cord arteriovenous malformation [SCAVM])
Fistula (spinal cord arteriovenous formation [SCAVF])
Macrofistula
Microfistula
Multifocal intradural lesions
- C.
According to location of SAVS
Intradural
Intramedullary
Extramedullary
Radicular
Filum
Epidural AVS (intraspinal)
Parachordal arteriovenous formation (AVF)
Branchial AVF
Vertebrovertebral arteriovenous formation (VVAVF)
Paraspinal arteriovenous formation (PSAVF)
The pathophysiology of spinal cord arteriovenous shunts (SCAVSs) is poorly understood. Like BAVMs, they are not diagnosed antenatally, although they are congenital . Most likely, a patient is born with a propensity to form an AVS, which then appears after a second or third event interferes with vessel repair or angiogenesis, although this process remains to be clarified. Presumably, etiologic factors important in the formation of BAVMs are also present in SAVSs. BAVMs are thought to represent populations of structurally unstable blood vessels with phenotypic evidence of upregulated angiogenesis . Animal models suggest that errors in artery-vein identity, as dictated by the Notch signaling pathway, may also play a role . Investigators have filled in some of the gaps in our knowledge of the pathophysiology of SAVSs with work done on the genetics, vascular biology, and endothelial cell biology of HHT in human beings and animal models.
Spinal cord arteriovenous lesions and hereditary hemorrhagic telangiectasia
HHT is an autosomal dominant disease with remarkably variable manifestations, including multifocal and multiorgan vascular dysplasias. Although classically defined by the triad of epistaxis, telangiectasias, and family history, patients with HHT can harbor AVSs in the pulmonary, hepatic, and CNS vasculature ( Box 2 , Tables 1 and 2 ) . This disease is also likely underdiagnosed, but its prevalence is estimated to be between 1 in 5000 to 1 in 8000 persons, with clusters of higher prevalence in some Caribbean islands, Denmark, and the Jura region in Central Europe . CNS involvement in this disease is common; up to 10% to 20% of individuals with HHT have BAVSs or SAVSs ( Fig. 2 ) .
Nosebleeds, spontaneous and recurrent (rare in children)
Telangiectasias, multiple, at characteristic sites, including lips, oral cavity, fingers, and nose (rare in children)
Visceral lesions
Gastrointestinal telangiectasia (with or without bleeding)
Pulmonary arteriovenous malformation (AVM)
Hepatic AVM
Cerebral AVM
Spinal AVM
Family history: first-degree relative with HHT according to these criteria
Molecular DNA sequencing for endoglin (ENG) and activin receptor-like kinase (ALK1)
Many symptoms and signs of HHT may not be present until late in life
If two criteria are met, a diagnosis is possible; if three or more criteria are met, a diagnosis is definite.
Modified from Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91:66–7; with permission.
| Age | <2 years | <16years |
|---|---|---|
| M:F ratio | 2:1 | 4:1 |
| Hemorrhage | 23% | 70% |
| Weakness | 62% | 73% |
| Bruit | 8% | NS |
| Sphincter | 15% | 27% |
| Asymptomatic | 8% | 17% |
| HHT | 46% | 17% |
| SAMS | 15% | 6% |
| Age at presentation | <2 years | <16 years |
|---|---|---|
| Mean no. treatment sessions | 2 | 1.5 |
| Cured | 78% | 20% |
| >50% occluded | 100% | 100% |
| Complications (minor) | 8% | 10% |
| Complications (major) | 0% | 0% |
| Improved | 75% | 80% |
| Stable | 25% | 20% |
| Worse | 0% | 0% |
Related posts:
 Endovascular Treatment of Hemangiomas
Endovascular Treatment of Hemangiomas
 Pediatric Neuroanesthesia
Cerebral Sinovenous Thrombosis in Children
Pediatric Neuroanesthesia
Cerebral Sinovenous Thrombosis in Children
 Overt and Incomplete (Silent) Cerebral Infarction in Sickle Cell Anemia: Diagnosis and Management
Overt and Incomplete (Silent) Cerebral Infarction in Sickle Cell Anemia: Diagnosis and Management
 Vein of Galen Aneurysmal Malformations
Current Endovascular Management of Maxillofacial Vascular Malformations
Vein of Galen Aneurysmal Malformations
Current Endovascular Management of Maxillofacial Vascular Malformations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


