(1)
Department of Clinical Radiology, Amiri Hospital – Kuwait City, Kuwait City, Kuwait
3.3.2 Acromegaly and Gigantism
3.3.4 Carney’s Complex
3.4 Osteoporosis
3.4.1 Primary Osteoporosis
3.4.2 Secondary Osteoporosis
3.6 Scurvy
3.7 Fluorosis
3.9.1 Cushing’s Syndrome
3.9.3 Addison’s Disease
3.9.4 Pheochromocytoma
3.9.5 Neuroblastoma
3.10.2 Precocious Puberty
3.10.4 Gynecomastia
3.10.5 Intersex Disorders
3.1 Graves’ Disease (Hyperthyroidism)
Graves’ disease (GD) is an autoimmune disorder characterized by hyperthyroidism, thyroid goiter, and ophthalmopathy. The disease arises due to the production of autoantibodies that auto-stimulates the thyrotropin receptors in the thyroid gland to secrete thyroid hormones.
GD clinical manifestations are mainly due to hyperthyroidism (thyrotoxicosis). Patients are commonly females between the third and fifth decades presenting with thyroid goiter. The thyroid is hypervascular, with venous humming that can be heard by stethoscope in some cases.
Systemic manifestations of hyperthyroidism include rapid weight loss (>10 % of body weight in less than 6 months), profuse sweating and heat intolerance, increased appetite (85 %), anorexia (15 %), increased bowel motion and diarrhea, oligomenorrhea in females, gynecomastia in males due to increased sex hormone-binding proteins, and proximal muscle weakness and muscle wasting due to increased basal metabolic rate. Skin manifestations include skin moisture due to sweating, vitiligo, and pretibial skin thickening due to mucin deposition in the dermis (myxoedema).
Graves’ ophthalmopathy is the most characteristic sign of this disease. GD is the most common cause of exophthalmos (abnormal prominent eye) and proptosis (protrusion) of globe in adults. It occurs in 35 % of cases. The proptosis can precede the actual thyroid abnormalities or occur after the disease has been brought under control. Proptoses are commonly bilateral and symmetrical; unilateral proptosis is uncommon.
Proptosis in GD can be explained by:
Infiltration and deposition of mucopolysaccharidosis (hyaluronic acid) into orbital muscles. The muscles’ bellies are characteristically increased in size, while their tendons are spared (fusiform enlargement). The inferior rectus and the medial rectus muscles are the most commonly involved. The lateral rectus is the last muscle to be involved. Hypertrophy of the lateral rectus only can be seen in orbital pseudotumor, and hypertrophy of the superior rectus only can be seen in orbital lymphoma.
Increased volume of the retrobulbar fat which will push the globe anteriorly.
Clinical signs of Graves’ ophthalmopathy include widened palpebral fissure (Dalrymple’s sign), staring expression with infrequent blinking (Stellwag’s sign), lid lag on downward gaze (von Graefe’s sign), and poor convergence (Möbius’s sign). Up to 5 % of patients with Graves’ ophthalmopathy develop optic neuropathy due to compression of the nerve in its canal because of backward herniation of the retro-orbital fat through the optic canal or from hypertrophied ocular muscle belly at the orbital apex.
Signs on US and Doppler Sonography
The gland is diffusely hypoechoic and enlarged in size.
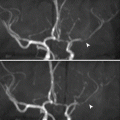
On color Doppler scan, the gland shows bilateral diffuse increase duplex signal due to hypervascularity. This sign is characteristic for GD and is called “thyroid inferno” sign (. Fig. 3.1.1).

Fig. 3.1.1
Color Doppler (Duplex) scan of the thyroid in a patient with Graves’ disease shows marked vascular signal due to bruit (thyroid inferno sign)
Signs of Graves’ Ophthalmopathy on CT and MRI
Bilateral, symmetrical increase in orbital muscles bellies width with spares tendons causing the orbital muscles to have fusiform appearance. The inferior rectus and the medial rectus muscles are characteristically affected (. Figs. 3.1.2 and 3.1.3).

Fig. 3.1.2
Axial ophthalmic CT image of a patient with Graves’ ophthalmopathy shows marked thickening of the medial rectus muscle of the left eye. Notice the difference in the medial rectus belly thickness (2) in comparison with the right eye (1)

Fig. 3.1.3
Coronal sinus and orbital CT illustration shows a differential diagnosis of recti muscles enlargement; the letter G stands for Grave’s disease, L for lymphoma, and P for orbital pseudotumor
Increases in the retrobulbar fat size.
CT evidence of proptosis is defined as globe protrusion exceeding the interzygomatic line by 21 mm or more on axial images at the level of the lens (. Fig. 3.1.4).

Fig. 3.1.4
Axial ophthalmic CT illustration demonstrates the interzygomatic line. A globe that protrudes >21 mm or more across this line is considered proptosis
GD optic neuropathy can be detected if retro-orbital fat is seen extending 4 mm beyond the boundary of the superior orbital fissure or if the optic nerve is seen compressed by a hypertrophied ocular muscle belly at the orbital apex.
Uncommonly, isolated dilatation of the superior ophthalmic vein may occur in patients with GD, and it can be easily mistaken for carotid–cavernous fistula. CT angiography can confirm the absence of carotid–cavernous fistula.
Further Reading
Arslan H et al. Power Doppler sonography in the diagnosis of Graves’ disease. Eur J Ultrasound. 2000;11:117–22.
Babcock DS. Thyroid disease in pediatric patient: emphasizing imaging with sonography. Pediatr Radiol. 2006;36:299–308.
Birchall D et al. Graves ophthalmopathy: intracranial fat prolapse on CT images as an indicator of optic nerve compression. Radiology. 1996;200:123–7.
Charkes ND et al. MR imaging in thyroid disorders: correlation of signal intensity with Graves disease activity. Radiology. 1987;164:491–4.
Greer MA et al. Hyperthyroidism. Dis Mon. 1967;13:1–45.
Nugent RA et al. Graves orbitopathy: correlation of CT and clinical findings. Radiology. 1990;177:657–82.
Ralls PW et al. Color-flow Doppler sonography in Graves disease: “thyroid inferno”. AJR Am J Roentgenol. 1988;150:781–4.
Rawson RW. Hyperthyroidism. Dis Mon. 1955;1:3–43.
Reed Larsen P. Hyperthyroidism. Dis Mon. 1976;22:1–30.
3.2 Hyperparathyroidism
Hyperparathyroidism is a metabolic disease characterized by the metabolic triad of high serum calcium level (hypercalcemia), low serum phosphorus level (hypophosphatemia), and increased calcium and phosphorus renal excretion (hypercalciuria).
Hyperparathyroidism can be caused by increased parathyroid hormone (PTH) release due to parathyroid adenoma or hyperplasia (primary type), chronic renal failure or parathyroid glands insensitivity to elevated serum calcium level (secondary type), or chronic renal failure with autonomous PTH release even after correction of the renal failure (tertiary type). Chronic renal failure causes reduction in serum calcium level, which induces hypersecretion of PTH to elevate serum calcium level. PTH increases serum calcium by increasing osteoclastic activity, promoting vitamin D renal hydroxylation, and promoting tubular renal absorption of calcium.
Hyperparathyroidism generally arises in those endocrine phases of life when endocrine glands are most active or rapidly changing like puberty, during the active phase of sexual life, or after menopause. Thus, hyperparathyroidism is rare before puberty and less commonly starts in later decades.
Symptoms and clinical presentation of hyperparathyroidism are related to its complications. Renal stone formation is one of the most common presentations of hyperparathyroidism. Increased renal excretion and serum calcium level promotes renal calculi formation. Peptic ulcers may occur in association with hyperparathyroidism for unknown reasons. It is speculated that changes in the calcium ion concentration may play a role in parasympathetic nervous system tone, which predisposes to increased secretions of gastric acids by increased vagal activity.
Episodes of acute pancreatitis are commonly associated with hyperparathyroidism for unknown reasons. Thirst and urinary frequency are common symptoms. Muscle fatigue and low back pain are also common complaints, and they are independent of bone changes.
The most common metabolic changes in hyperparathyroidism are observed in the skeletal system. Diffuse osteoporosis and bone resorption are commonly seen in primary hyperparathyroidism. In contrast, diffuse or focal osteosclerosis is observed in secondary hyperparathyroidism. Subperiosteal, subchondral, and subligamentous bone resorptions are the most common findings radiologically.
Brown tumor is an eccentrically located, expansile bony lesion uncommonly seen in secondary hyperparathyroidism. In severe hyperparathyroidism, large areas of bone marrow cavity are lost due to bone resorption. This bony resorption leads to microfractures and bleeding in the resorbed areas, which will create a mass-like effect within the trabecular bone. This mass-like structure has a brown pigment in gross section due to hemosiderin content. Gradually, this mass undergoes cystic changes. As the severity of the disease increases, these changes can progress to severe and diffuse type of bone expansion, cystic changes, and bone marrow fibrosis, a condition which is known as osteitis fibrosa cystica. Brown tumor mimics giant cell tumor (osteoclastoma) radiologically and histologically. Differentiation between the two clinical conditions depends on the presence or absence of hyperparathyroidism manifestations. Osteitis fibrosa cystica is a rare complication of hyperparathyroidism that is seen in advanced stage disease. It is usually seen in young patients <20 years.
Nephrocalcinosis is a condition characterized by calcification and calcium deposition within the renal parenchyma, either in the cortex or in the medulla. Cortical nephrocalcinosis occurs due to prior insult to the renal cortex like in tuberculosis, ischemia, and glomerulonephritis. Usually it affects one kidney, and the affected kidney is small with global atrophy. Medullary nephrocalcinosis, on the other hand, arises due to calcification of the medullary pyramids due to deposition of calcium within the renal tubules. Medullary nephrocalcinosis is the most common type of nephrocalcinosis (95 %) and is caused by systemic hypercalcemic states like in hyperparathyroidism, distal renal tubular acidosis, malignancy, and acute sarcoidosis. Typically, it affects both kidneys in a bilateral and symmetrical fashion, because the cause usually is a systemic disease.
Primary hyperparathyroidism can be a part of multiple endocrine neoplasia (MEN) syndrome. MEN syndrome is characterized by the occurrence of tumors involving two or more endocrine glands within a single patient. There are two major types of MEN: MEN type 1 (MEN1, Wermer’s syndrome) and MEN type 2 (MEN2, Sipple’s syndrome). Both syndromes are inherited as autosomal dominant. MEN1 is characterized by the combined occurrence of parathyroid tumors, pancreatic islet cell tumors (e.g., gastrinoma), and anterior pituitary tumors (e.g., prolactinoma). Associated tumors include adrenal tumors, carcinoid tumors, and lipoma. Although not part of the original description, meningioma has been reported to occur in patients with hyperparathyroidism due to MEN type 1. MEN type 2, on the other hand, is divided into three subtypes: MEN2a, MEN2b, and MTC only. MEN2a describes the association of medullary thyroid carcinoma (MTC), pheochromocytoma, and parathyroid tumors. MEN2b describes the association of MTC, pheochromocytoma, marfanoid body habitus, mucosal neuromas, and megacolon. Lastly, MTC only is a variant in which MTC is the sole manifestation of this syndrome.
In up to 2 % of normal people, an ectopic parathyroid tissue may be found within the mediastinum. The ectopic parathyroid tissue is commonly located within the anterior mediastinum. An ectopic parathyroid adenoma is rare and should be suspected in a patient with hyperparathyroidism who was operated and the signs and symptoms of hyperparathyroidism persisted (5–10 % of cases). Other areas where ectopic parathyroid tissue may be found include the neck (45 %), upper cervical area (8 %), or along the aortic arch (5 %).
Differential Diagnoses and Related Diseases
Hyperparathyroidism–jaw tumor syndrome is a rare, autosomal recessive disease characterized by hyperparathyroidism (90 %), ossifying fibroma of the maxilla and/or mandible (30 %), renal cysts and/or tumors (10 %), and uterine tumors. Ossifying fibroma is a benign lesion that arises from cells in the periodontal ligament and is mainly restricted to the tooth-bearing areas of the jaw. The lesion is visualized as a well-demarcated bony lesion composed of fibrocellular tissue and mineralized material. The tumor is typically painless and located at the posterior region of the mandible. Patients are often >35 years old. However, a juvenile form (<20 years) may be seen.
Hungry bone syndrome (HBS) is a rare complication of parathyroidectomy manifested by severe, prolonged, sometimes life-threatening hypocalcemia. The hypercalcemia in hyperparathyroidism is mainly due to increased bone turnover with predominant osteoclastic bone resorption and increased renal tubular absorption of calcium. After parathyroidectomy, the PTH stimulus over the osteoclasts is suddenly removed, stopping the osteoclastic activity, but the osteoblastic activity continues at its high rate, resulting in marked increase in bone uptake of calcium to facilitate bone remodeling. The excessive osteoblastic bony remodeling causes severe hypocalcemia. HBS is seen in 12 % of parathyroidectomy cases, and it is suspected in patients who had parathyroidectomy and presented with persistent hypocalcemia and hypophosphatemia. Predisposing factors for HBS include parathyroid adenoma >5 cm in diameter, high preoperative PTH, calcium, and alkaline phosphatase levels, advanced age, and osteitis fibrosa cystica.
Signs on Plain Radiographs
On chest radiograph, tracheal shift due to enlarged parathyroid adenoma may be the first sign detected in an asymptomatic patient.
On abdominal radiographs, urinary tract calcium calculi are seen as radio-opaque lesions in the renal area or the urethral course.
Cortical nephrocalcinosis is often detected as a unilateral renal “eggshell calcification,” while medullary nephrocalcinosis is detected as multiple, punctuated calcification seen within the kidney shadows in a bilateral symmetrical fashion (. Fig. 3.2.1).

Fig. 3.2.1
A plain radiograph of the kidneys in a patient with medullary nephrocalcinosis shows bilateral, almost symmetrical, punctuated calcification within the renal shadow
Signs on Skeletal Radiographs
Diffuse osteoporosis and lytic bony lesions are commonly found in primary hyperparathyroidism.
Widening of sacroiliac joints due to subchondral bone resorption can be seen.
Salt and pepper skull appearance: this occurs due to resorption of the trabecular bone in the skull and replacement of the resorbed bone by a newly formed connective tissue causing loss of integrity in the shape of the skull bones (. Fig. 3.2.2).

Fig. 3.2.2
A lateral plain radiograph of the skull shows mild salt and pepper skull lesions in a patient with primary hyperparathyroidism
The vertebral bodies in secondary hyperparathyroidism show sclerosis of the end plates (Rugger–Jersey spines) (. Fig. 3.2.3).

Fig. 3.2.3
A lateral spine radiograph of a patient with secondary hyperparathyroidism shows diffuse vertebral end plate sclerosis (Rugger–Jersey spines)
Subperiosteal cortical resorption typically occurs in the hand, especially at the radial aspect of the middle phalanx, which is a specific sign seen in both primary and secondary hyperparathyroidism (. Fig. 3.2.4).

Fig. 3.2.4
A plain radiograph of the fingers shows radial side subperiosteal resorption of the middle and distal phalanges (arrowheads), a specific sign of prolonged hyperparathyroidism
Brown tumor is seen as a well-circumscribed cystic bony lesion which can cause bone expansion. There are often multiple lytic lesions found together. When the hyperparathyroidism is treated, the brown tumor undertows ossification and will transform into a bone island (sclerotic lesion). The most common areas for brown tumors are the pelvis, rib, long bone diaphysis, clavicle, and mandible (. Fig. 3.2.5).

Fig. 3.2.5
A femoral diaphyseal lytic, expansile bony lesion in a patient with prolonged hyperparathyroidism. Pathological biopsy proved to be brown tumor
Subligamentous bone resorption at the sites of ligament insertion into bone can be seen. It is commonly observed at the elbows over the olecranon, plantar aspect of the calcaneus, and the superior pole of the dorsal aspect of the patella.
Chondrocalcinosis occurs due to deposition of calcium pyrophosphate dehydrate into the cartilage of the joints (metastatic calcification). It is found in up to 40 % of hyperparathyroidism cases.
Osteitis fibrosa cystica presents as a lytic expansile bony lesion that mimics metastatic bone disease (. Fig. 3.2.6).

Fig. 3.2.6
A plain hip radiograph of a patient with prolonged undiagnosed hyperparathyroidism. The right side of the hip shows numerous bony lytic and sclerotic lesions with a semi-moth-eating appearance that was initially thought to be Paget’s disease. Bone biopsy proved to be osteitis fibrosa cystica
Diffuse osteosclerosis is commonly seen in patients with secondary hyperparathyroidism (. Fig. 3.2.7).

Fig. 3.2.7
A plain abdominal radiograph of a patient with secondary hyperparathyroidism shows diffuse osteosclerosis
Signs on US
Thyroid ultrasound often shows oval or round hypoechoic mass in the posterior inferior poles of the thyroid (usually <3 cm in diameter) representing parathyroid adenomas. The mass has a well-defined echogenic line separating the adenoma from the thyroid gland representing the capsule. The mass shows internal cystic changes, mixed echogenicity, or calcification as the size exceeds 3 cm in diameter.
Renal calculi are seen as hyperechoic lesions with posterior shadowing.
Medullary nephrocalcinosis is detected as hyperechogenic renal pyramids.
Signs on Doppler Sonography and PD
The parathyroid adenoma typically shows high blood flow signal and perfusion, especially at the peripheral portion of the adenoma.
Signs on CT and MRI
On CT, parathyroid adenoma is detected as a well-defined mass located in the posterior/inferior pole of the thyroid with intense enhancement after contrast administration. On MRI, the mass shows intermediate T1 and high T2 signal intensities with intense enhancement after contrast injection.
Ectopic parathyroid adenoma is identified as an anterior mediastinal mass with high contrast enhancement (similar to the usual parathyroid adenomas). The ectopic parathyroid mediastinal adenoma is classically <2 cm in diameter.
Brown tumors have characteristically low T2 signal intensity due to hemosiderin content. It shows early intense enhancement after contrast injection due to marked vascularity. Fluid–fluid levels may be observed within the tumors in some cases due to intramural bleeding.
Ossifying fibroma is seen on CT as a well-demarcated lytic lesion with mixed mineralized material (up to 50 % are purely lytic lesions). The lytic lesion typically is expansile and may mimic fibrous dysplasia with its ground-glass appearance if the matrix is extensively calcified (. Fig. 3.2.8). On MRI, ossifying fibroma typically shows low to intermediate signal intensity on both T1W and T2W images with homogeneous contrast enhancement after contrast injection.

Fig. 3.2.8
A CBCT dental illustration shows ossifying fibroma as lytic expansile bony lesion with mixed calcified matrix
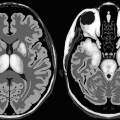
The salt and pepper skull appearance seen in plain radiograph can be seen on MRI as bone resorption (. Fig. 3.2.9).

Fig. 3.2.9
Two different patients with T1W image MRI of the brain. In image (a), there is marked bone resorption of the inner surface of the skull in a patient with prolonged primary hyperparathyroidism (salt and pepper skull appearance). Compare the skull bones with the normal skull in image (b)
Further Reading
Ahuja AT et al. Imaging of primary hyperparathyroidism – what beginners should know. Clin Radiol. 2004;59:967–76.
Bertolini F et al. Multiple ossifying fibromas of the jaw: a case report. J Oral Maxillofac Surg. 2002;60:225–9.
Eggert P et al. Nephrocalcinosis in three siblings with idiopathic hypercalciuria. Pediatr Nephrol. 1998;12:144–6.
Falchetti A et al. Multiple endocrine neoplasia type I variants and phenotypes: more than nosological issue. J Clin Endocrinol Metab. 2009;94:1518–20.
Ghanaat F et al. Hungry bone syndrome: a case report and review of the literature. Nutr Res. 2004;24:633–8.
Hsieh M-C et al. Pathologic fracture of the distal femur in osteitis fibrosa cystica simulating metastatic disease. Arch Orthop Trauma Surg. 2004;124:489–501.
Kabala JE. Computed tomography and magnetic resonance imaging in diseases of the thyroid and the parathyroid. Eur J Radiol. 2008;66:480–92.
Krudy AG et al. The detection of mediastinal parathyroid glands by computed tomography, selective arteriography, and venous sampling. Radiology. 1981;140:739–44.
McDonald DK et al. Primary hyperparathyroidism due to parathyroid adenoma. Radiographics. 2005;25:829–34.
Reuter K et al. Unsuspected medullary nephrocalcinosis from furosemide administration: sonographic evaluation. J Clin Ultrasound. 1985;13:357–9.
Rypins EL. Osteitis fibrosa cystica at unusual age. J Bone Joint Surg Am. 1933;15:509–12.
Schmidt BP et al. Hyperparathyroidism-jaw tumor syndrome: a case report. J Oral Maxillofac Surg. 2009;67:423–7.
Smith D et al. Hungry bones without hypocalcemia following parathyroidectomy. J Bone Miner Metab. 2005;23:514–5.
Takeshita T et al. Brown tumor with fluid-fluid levels in a patient with primary hyperparathyroidism: radiological findings. Radiat Med. 2006;24:631–4.
Wang Q et al. Power Doppler imaging findings in multilocular giant parathyroid adenoma which caused hypercalcaemic crisis. J Laryngol Otol. 1998;112:769–99.
3.3 Growth Hormone Diseases
The human growth hormone (GH) is a polypeptide consisting of 188 amino acids and having a molecular weight of 21,500. GH from other species shares partial sequences of amino acids in common with human GH (e.g., bovine GH). These partial sequences consist of active cores, which are pharmacologically active. Thus, it may be not necessary to synthesize the entire bovine GH molecule to yield an actively working substance in humans.
GH disorders result from either excess or reduction of its secretion within the body. The normal GH is secreted in two cyclic rhythms: one in the morning and the other in the evening.
Growth Hormone Insufficiency (Hypopituitarism)
GH insufficiency (hypopituitarism) can be idiopathic (primary) or due to pituitary gland tumor (secondary). Idiopathic GH insufficiency children exhibit growth retardation, delayed puberty, and hypothyroidism without elevated thyroid-stimulating hormone (TSH) level. Growth retardation is assumed if the child falls more than three standard deviations below the mean for his/her age and also if the child’s growth rate is <50 % of the anticipated growth rate over a period of 1 year.
Pituitary stalk interruption syndrome (PSIS) is a form of GH insufficiency due to abnormal pituitary stalk. Children with PSIS have pronounced GH insufficiency, with or without other anterior pituitary hormonal deficiencies.
Signs on Skeletal Radiographs
Plain skeletal radiographs can be used to accurately assess bone age according to the bone maturation. Each bone in the body starts to ossify at a certain age. By imaging certain bones within the body, assessing their ossification maturation, and comparing it to a standard reference of bone maturation of the patient’s current age, the radiologist can easily assess the patient bone maturation rate. This method is a valuable tool that can detect GH abnormalities in a relatively short time with much accuracy.
Both hands and elbows are often X-rayed, and the shapes of all epiphyses of the radius, ulna, carpals, metacarpals, and all the phalanges are assessed in comparison with a standard reference. Delayed bone maturation can be seen in GH insufficiency and hypothyroidism.
The normal appearance of primary ossification centers of the wrist: capitate (2–3 months), hamate (3 months), triquetral (2–3 years), lunate (3 years), trapezium (3–4 years), trapezoid (4 years), scaphoid (4–5 years), pisiform (8–9 years), ulnar epiphysis (6–7 years), and radial epiphysis (1 year).
The normal appearance of primary ossification centers of the elbow (CRITOE): capitulum (6 months), radial head (5 years), internal (ulnar) epicondyle (6–7 years), trochlea (9 years), olecranon (9–10 years), and external (radial) epicondyle (10–11 years).
By applying the previous primary ossification centers age to a skeletal radiograph of a child, radiologists can estimate roughly the age of that child. However, precise age estimation should be assessed using a standard reference (. Fig. 3.3.1).

Fig. 3.3.1
A plain radiograph of the hand, wrist, and forearm in a 4-year-old boy with growth retardation shows skeletal maturation retardation. Although the child’s age is 4 years, only the capitate and hamate bones are ossified (arrowhead), which commonly start ossification at 2–3 months. At 4 years of age, we expect the scaphoid, lunate, and trapezium to be seen too. Moreover, the elbow shows only the ossification center of the capitulum (arrow), which starts to ossify at 6 months of age. It seems as if the patient’s age has been stunted at 6–12 months old
Signs on MRI
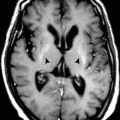
The pituitary on MRI in patients with GH insufficiency shows absence or marked thinning of the pituitary stalk, reduced size of the anterior pituitary, lack of the normal posterior pituitary high signal, and presence of a high signal nodule in the region of the infundibular recess of the third ventricle representing ectopic posterior pituitary (. Fig. 3.3.2).

Fig. 3.3.2
Sagittal T1W sella MR illustration demonstrates interruption of the pituitary stalk with ectopic high T2 signal intensity characteristic of ectopic neurohypophysis and pituitary stalk interruption syndrome (PSIS) (arrowhead)
Acromegaly and Gigantism
Excess GH pituitary release or GH abuse in body builders results in two disorders named acromegaly and gigantism. The term acromegaly was used for the first time by “Pierre Marie” in France in 1886 describing patients with characteristic hands and feet (acro) hypertrophy (megaly). Acromegaly literally means hypertrophy of the extremities. The disease has been described in historical writings, especially in people whose body development is considerably greater than normal and who are looked upon as giants. It is even described in the Jewish Talmud by the Biblical name sarua that refers to abnormal growth of a single limb, which rendered a priest unfit to serve in the temple.
Acromegaly is an adult disease characterized by increased production of the GH resulting in characteristic body changes. When the excess GH release starts in adolescence with open epiphyses, the condition is called “gigantism.” The common etiology in both cases is a pituitary adenoma that increases GH production. GH causes retention of nitrogen with an overall anabolic effect. It also increases the transport of amino acids in the tissues and their release into proteins and mobilizes lipids from adipose tissue increasing their oxidation as a source of energy, thus sparing muscle glycogen. Due to previous GH effects, an athlete who abuses GH may realize an improvement in performance and strength with the use of GH supplements. Amino acid supplements of arginine, ornithine, and lysine, in combination or alone, can stimulate the production of endogenous GH. GH release can be also stimulated by some medications like l-dopa, clonidine, and propranolol. Athletes with GH abuse may develop acromegaly or acromegalic-like state with complications similar to acromegaly.
Patients with acromegaly are characterized by overgrowth of the terminal parts of the skeleton (e.g., hands) and the soft-tissue parts of the viscera. The earliest complaints include headache, visual defects in 30 % of patients (bitemporal), fatigability, asthenia, and sweating. Later, the size of the head, hands, and feet starts to grow progressively.
Patients with acromegaly develop characteristic appearance. The facial features become coarse and thickened, with enlargement of the nose. Protrusion of the mandible (prognathism), tongue enlargement, widening of the teeth, vertebral kyphosis, skin thickening, and protrusion of the supraorbital ridges are also characteristic features. The heart, spleen, liver, and kidneys may be enlarged. Patients with acromegaly show higher tendency toward gastrointestinal cancers (e.g., colon cancer).
Women with acromegaly show high incidence of intrauterine bleeding or amenorrhea. In both males and females, there is gradual loss of libido, and testicular or ovarian atrophy may develop later in life.
Other hormonal abnormalities may be found in patients with acromegaly. Thyroid enlargement usually occurs due to hypertrophy with increased thyroid function rate. Inappropriate lactation due to hyperprolactinemia may be found in some patients. Increased serum phosphorus level is a characteristic feature of acromegaly due to increased tubular reabsorption. Adrenal gland hypertrophy without signs of cortical hyperfunction may occur. Lastly, large patients with acromegaly develop diabetes mellitus due to the diabetogenic effect of GH which increases the serum blood glucose level. Most of the hormones in the body increase the serum glucose blood level like glucagons, cortisol, and GH. Only insulin is capable of reducing the serum glucose blood level.
Patients with gigantism exhibit the same clinical and radiological features as acromegaly. The characteristic feature in gigantism is the tall height of the patients. Increased GH production delays the closure of the epiphyses, so patients will start to grow in height beyond the normal age of epiphyseal closure. This may result in patients reaching a height of up to 2.4 m according to the historical medical literature.
Differential Diagnoses and Related Diseases
Van Buchem disease is a rare hereditary disorder characterized by endosteal hyperostosis of the skull and the mandible due to excessive lamellar bone deposition with narrow Haversian canals. The disease has both autosomal dominant and recessive forms. Clinically, van Buchem disease may resemble acromegaly but not radiologically. Patients with van Buchem disease present with thickening of the bridge of the nose, deafness due to petrous bone thickening, eye abnormalities due to stenosis of the optic canal, and cranial nerve palsies due to hyperostosis at the base of the skull (. Fig. 3.3.3).

Fig. 3.3.3
An illustration of the skull in a lateral view shows the gross pathological changes seen in van Buchem disease. Notice the enlargement and thickening of the mandible, with sclerosis of the skull base and calvarium
Signs on Skeletal Radiographs
Increased thickness of the flat bones of the skull (. Fig. 3.3.4).

Fig. 3.3.4
A lateral skull radiograph of a patient with acromegaly shows thickened occipital bone (arrowheads) and enlargement of the frontal and maxillary sinuses (arrows)
Enlargement of the sinuses and mastoid air cells (. Fig. 3.3.4).
Enlargement of the external occipital protuberance (. Fig. 3.3.4).
Enlargement of the sella turcica (due to adenoma). The posterior clinoid processes may show signs of erosions.
Enlargement of the vertebrae, especially in their transverse diameter.
Increased size and widening of the distal phalangeal tufts (spade-like appearance) (. Fig. 3.3.5).

Fig. 3.3.5
Anteroposterior plain radiograph of the third and fourth fingers of a patient with acromegaly shows widening of the distal phalangeal tufts (spade-like appearance)
Increased soft-tissue thickening of the heel pad (normal up to 23 mm in men and 21 mm in women).
Hypertrophic osteoarthritis of the joints.
Locking of the metacarpals is a relatively rare condition that can be seen in patients with acromegaly. The condition is characterized by a hooklike osteophytes formation in the heads of the metacarpal bones. As the patient makes a fist or grasps something, the volar distal part of the proximal phalanx will be locked against the osteophyte in the metacarpal head, locking the finger in the grasping position.
Hyperostosis frontalis interna is a condition where thickening of the inner surface of the frontal bone may be seen in some cases with acromegaly.
In van Buchem disease, there is generalized skull hyperostosis, mandibular hyperostosis and enlargement, ribs and clavicular thickening, and diaphyseal endosteal sclerosis that spares the bone ends, especially in the phalanges. Unlike acromegaly, there is no dental widening or mandibular prognathism.
Signs on Brain MRI
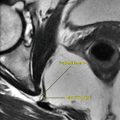
Pituitary adenoma is seen as a bulging lesion in the superior or inferior aspect of the pituitary gland with low T1/high T2 signal. The adenoma is hypointense compared to the normal pituitary tissue on postcontrast images (. Fig. 3.3.6).

Fig. 3.3.6
Sagittal T1W (a) and T1Wpostcontrast (b) sella MRI in a patient presented with features of acromegaly. The MR examination showed macroadenoma with cystic changes. The adenoma is detected as a mass with low contrast enhancement (arrowhead) compared with the highly enhanced normal pituitary tissue due to its rich blood supply
Indirect signs of pituitary adenoma include convex upper border of the gland with shifted pituitary stalk. When the cavernous internal carotid artery is completely surrounded by the tumor, then the cavernous sinus is mostly invaded by the tumor.
Growth Hormone Insensitivity (Laron Syndrome)
Laron syndrome (LS) is a rare, autosomal recessive, congenital disease characterized by GH receptors gene defects, resulting in lack of body tissue response to GH.
Patients with LS present with dwarfism, severe growth retardation, and characteristic facial features. Most cases are reported from patients with Oriental Jewish origin or patients originating in the Mediterranean area like Arab, Turkish, Iranian, and Pakistani origin.
Patients with LS typically have small chin (micrognathia), underdeveloped facial bones, smaller head circumference according to age, protruding forehead, and saddle nose deformity due to nasal bone underdevelopment. The teeth are defective and crowded due to micrognathia. The hair is silky and shows frontal and temporal thinning. Alopecia is often seen in males (. Fig. 3.3.7).

Fig. 3.3.7
An illustration of a child demonstrates the characteristic features of Laron syndrome (LS) like micrognathia, silky hair with temporal thinning, saddle nose deformity, and mildly protruding forehead
Patients are usually obese due to underdevelopment of bones and muscles. The children and even adults have very high-pitched voices due to narrow oropharynx. Hands and feet are small (acromicria). The genitalia and gonads are small since birth, and males show delayed puberty more than females. LS patients do not have real pubertal growth spurt.
Laboratory investigations show severe hypoglycemia in neonates that improves with age, low serum alkaline phosphatase and creatinine, low serum cholesterol, and low-density lipoproteins.
Hormonal investigations show increased serum GH levels with very low serum levels of insulin-like growth factor-I (IGF-I). IGF-I is the anabolic effector hormone of GH. Prolactin levels may be elevated due to a drift phenomenon to the GH secretion. Serum insulin level is usually high with hypoglycemia.
Signs on Radiographs
Generalized bone maturation delay and osteoporosis.
Epiphyseal closure occurs after age 16–18 in girls and 20–22 in boys.
Underdeveloped facial bones, with thin diploe of the skull.
Atlantoaxial joint degeneration and spinal stenosis is often observed.
Os odontoideum may be seen. Os odontoideum is a situation where the axial den (odontoid process) is hypoplastic, absent, or separated from the axis body as a congenital variant (not due to previous trauma). It is due to failure of the three dens ossification centers to fuse together with the axis body. It is seen as a round ossicle with smooth edges over the axis body in open mouth view (best view to evaluate the dens). It may be impossible to differentiate os odontoideum from a previous old dens fracture without history.
Carney’s Complex
Carney’s complex (CNC) is a rare disease characterized by the formation of multiple endocrine and nonendocrine tumors, spotty skin pigmentation, myxomas, and endocrine overactivity. The disease is also known as NAME syndrome (nevi, atrial myxoma, myxoid neurofibromata, and freckles) and LAMB syndrome (lentigines, atrial myxoma, mucocutaneous myxomas, and blue nevi).
CNC condition has an autosomal dominant mode of inheritance. Carney’s syndrome is a different clinical condition characterized by a triad of several neoplasms including gastric epithelioid leiomyosarcoma, pulmonary chondroma, and extra-adrenal paraganglioma. Patients with CNC are diagnosed by fulfilling two or more of the CNC diagnostic criteria.
Carney’s Complex Major Diagnostic Criteria
Lentiginosis and blue nevi: Lentigo is a brownish-black flat macule that is typically found in the lips, around the inner canthus of the eye, axilla, or genitals (. Fig. 3.3.8). When the macules are found diffusely in the body, the condition is called “lentiginosis.” The other characteristic skin lesion found in CNC is blue skin nevi.

Fig. 3.3.8
An eye and lip illustration demonstrates the lentigo pigmented lesions found in the inner canthus and the lips in a patient with Carney’s complex (CNS)
Cutaneous myxomas: These are seen on the trunk as small red papules.
Cardiac myxoma: It is the most common component of CNC. Cardiac myxoma is a gelatinous tumor, and it is the most common primary cardiac neoplasm in adults (50 % of cardiac neoplasms). Ninety percent of cases are seen in adult women between 30 and 60 years of age. Most cases are sporadic. Patients usually present with CNS symptoms, fatigue, arthralgia, fever, anemia, and weight loss. Twenty percent of myxomas are asymptomatic. Patients with CNC cardiac myxoma are younger than patients with sporadic myxoma (an average age of 24 years).
Acromegaly: CNC patients can develop acromegaly due to GH-releasing pituitary micro-/macroadenoma.
Primary pigmented nodular adrenocortical disease (PPNAD): It is a rare disease of children and young adults below 20 years of age. It can be the first manifestation of CNC. Pathologically, the adrenal shows small black, brown, red, or yellow nodules separated by atrophic adrenal cortex. Diagnosis is essentially based on histological findings. PPNAD is one of the common causes of ACTH-independent Cushing’s syndrome.
Large-cell calcifying Sertoli cell tumor (LCCSCT): Bilateral germ cell tumors of the testes are the initial presentation of CNC in 20 % of cases. Diagnosis is strengthened by the detection of high serum levels of estrogen or androgen.
One or more of the following cancers: breast fibromyxomas (25 % of cases), osteochondromyxoma, follicular thyroid carcinoma, and psammomatous melanotic schwannoma.
Signs on US
In LCCSCT, the testes show multiple, round, well-defined, large (5–10 mm) echogenic calcification with acoustic shadowing representing the stromal tumors.
Signs on MRI
Cardiac myxoma: myxoma typically appears as a heterogeneous mass on T2W images with a narrow-based attachment located in the interatrial septum at the area of fossa ovalis (90 % of cases). Eighty percent of myxomas arise in the left atrium and 10 % in the right atrium. Calcification is frequently seen, and the mass shows heterogeneous contrast enhancement. The location of the tumors is very characteristic.
LCCSCT are detected as multiple high T2 intratesticular masses with hypointense areas representing calcifications (. Fig. 3.3.9).

Fig. 3.3.9
Coronal T2W testicular MR illustration shows bilateral hypointense T2 signal intensity lesions in the testes bilaterally representing large-cell calcifying Sertoli cell tumor (LCCSCT)
Sella MRI may show pituitary adenoma especially in patients with signs of acromegaly.
PPNAD: the adrenal glands may be normal or show limbs macronodularity (>5 mm in size).
Further Reading
Boikos SA et al. Pituitary pathology in patients with carney complex: growth-hormone producing hyperplasia or tumors and their association with other abnormalities. Pituitary. 2006;9:203–9.
Cazabat L et al. PRKAR1A mutations in primary pigmented nodular adrenocortical disease. Pituitary. 2006;9:211–9.
Chakraborty PP et al. Laron’s syndrome in two siblings. Indian J Pediatr. 2007;74:870–1.
Daughaday WH et al. The pituitary in disorders of growth. Dis Mon. 1962;8:1–47.
Doppman JL et al. Cushing syndrome due to primary pigmented nodular adrenocortical disease: findings at CT and MR imaging. Radiology. 1989;172:415–20.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree