Fig. 9.1
A 17-year-old female with appendiceal carcinoid. Transverse ultrasound shows an enlarged appendix (arrows) with surrounding echogenic fat. No discrete mass was identified even though at resection a 5 mm carcinoid was present in the mid portion of the appendix
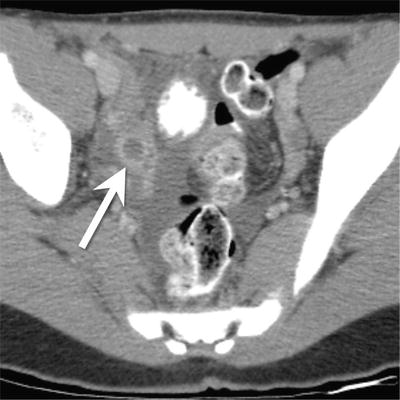
Fig. 9.2
A 12-year-old female with appendiceal carcinoid. Axial contrast enhanced CT shows a dilated appendix (arrow) with a mildly thickened and hyper-enhancing wall. There is a considerable amount of surrounding free fluid. No discrete mass was identified in the appendix by imaging even though at resection a 10 mm carcinoid was present
Ultrasound: Because of the small size of most appendiceal NETs, they are not often seen by ultrasonography. Larger tumors appear as asymmetric appendiceal wall thickening. Tumors with calcifications may appear as an echogenic focus with posterior acoustic shadowing [11], but these lesions can be difficult to distinguish from fecaliths.
CT: Due to its small size and location at the tip of the appendix, an appendiceal NET is rarely identified on preoperative CT. If it is seen, the tumor appears as diffuse wall thickening. Occasionally calcification within the tumor mimics an appendicolith [11]. CT can also be used to identify metastatic disease in lymph nodes and elsewhere. An irregular soft issue mass near the root of the mesentery is characteristic of metastatic NET [11].
Nuclear medicine: Both Indium-111 octreotide and Iodine-131 meta-iodobenzylguanidine can be used to identify metastatic sites of disease.
Pathology
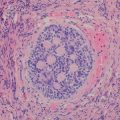
Gross and microscopic features: Grossly, gastrointestinal NETs form well circumscribed round lesions in the submucosa or muscular layer. The cut surface appears red to tan, reflecting an abundant microvasculature, or sometimes yellow because of high lipid content. In the majority of cases, the tumor is located at the tip of the appendix and is <1 cm in diameter (Fig. 9.3a). Tumor size (>2.0 cm) has been correlated with nodal metastasis but not with poor outcome [12].
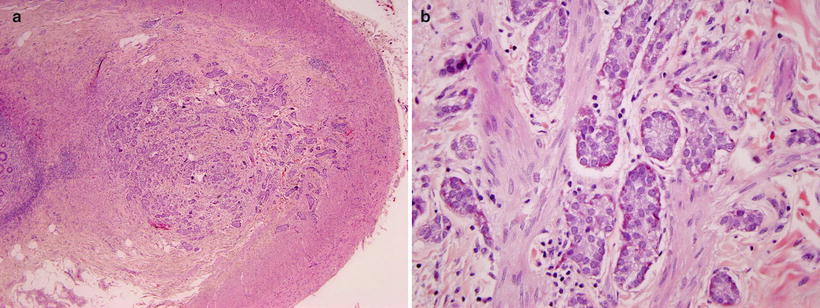
Fig. 9.3
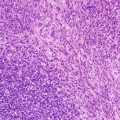
Appendiceal carcinoid. (a) A poorly circumscribed, non-encapsulated neoplasm arises from the tip of the appendix and invades the muscularis propria. (b) The lesion contains nest of neuroendocrine cells invading smooth muscle. The cells contain basal nuclei and pink granules
Morphologically, well-differentiated NETs display a characteristic “organoid” arrangement, with solid/nesting, trabecular, or gyriform patterns (Fig. 9.3b). The cells are relatively uniform and contain round to oval nuclei, coarsely stippled chromatin, and finely granular cytoplasm.
The College of American Pathologists (CAP) protocol grades well-differentiated appendiceal NETs as either G1 [<2 mitoses/10 high power fields (HPF)] or G2 (2–10 mitoses/10 HPF) [13–16]. G1 tumors represent “carcinoids,” and G2 tumors are “atypical carcinoids.” Tumors with >10 mitoses/10 HPF comprise poorly differentiated neuroendocrine carcinomas.
Immunohistochemistry and other special stains: NET cells produce abundant neurosecretory granules, reflected by strong and diffuse immunohistochemical expression of neuroendocrine markers such as synaptophysin and chromogranin. Some tumors may secrete specific peptide hormones or bioamines such as insulin, glucagon, somatostatin, vasoactive intestinal peptide (VIP), serotonin, and gastrin, which may produce clinically evident hormonal syndromes.
Molecular diagnostic features and cytogenetics: There are limited data about molecular genetics of appendiceal carcinoids.
Prognosis
Because most appendiceal NETs are incidentally discovered, they are initially treated with surgical resection. The size of the tumor is a key indicator of survival. Patients with tumors <2 cm have an overall excellent survival, whereas patients with tumors >3 cm have a significantly worse long-term outcome [17]. Other factors that lead to decreased survival include lymph node metastases and moderate to poor tumor cell differentiation [17].
Gastrointestinal Stromal Tumor (GIST)
Clinical Features and Epidemiology
Gastrointestinal stromal tumors are the most common mesenchymal tumor of the gastrointestinal tract in adults but are rare in children [18]. Isolated institutional reports estimate that pediatric GISTs account for about 2 % of all non-rhabdomyosarcomatous soft tissue sarcomas and 2 % of all GISTs [19, 20].
Pediatric GIST occurs with a median age at presentation of approximately 13 years with females representing over 70 % of cases [21, 22]. The vast majority occur in the stomach. Multifocal gastric tumors and lymph node metastases are more common in children [23].
While pediatric patients with GIST can be asymptomatic, they usually present with signs and symptoms of chronic GI bleeding such as anemia, weakness, or syncope [23]. Other possible symptoms include acute abdominal pain, bowel obstruction, or abdominal distension.
Imaging Features (Figs. 9.4 and 9.5)
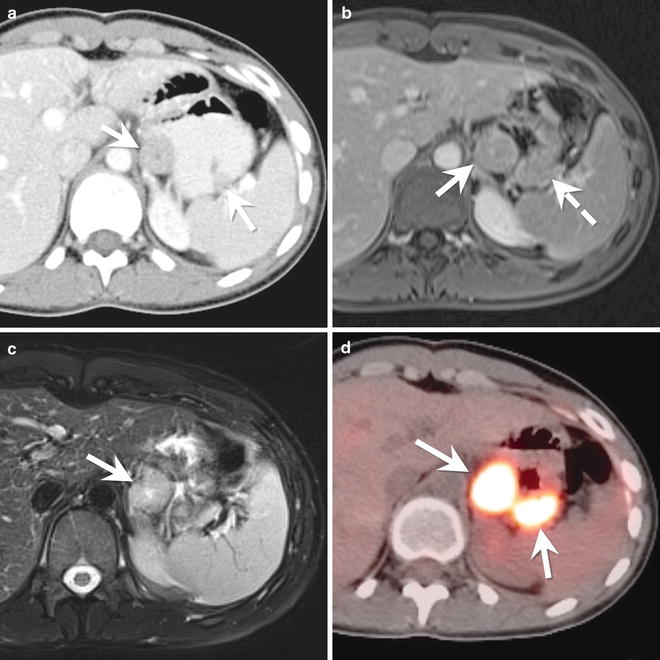
Fig. 9.4
A 14-year-old female with a gastrointestinal stromal tumor (GIST). (a) Axial contrast enhanced CT of the abdomen shows two exophytic masses (arrows) arising from the gastric antrum. The masses enhance less than the liver but more than the paraspinal musculature. (b) Axial T1-W post-contrast MR image of the upper abdomen shows the dominant mass (arrow) arising from the gastric antrum. The mass is isointense to the nearby liver. The smaller mass (dashed arrow) is not visible as a discrete mass on the MRI but instead appears as an area of gastric thickening. (c) On the axial T2-W MR image, the dominant mass (arrow) is hyperintense compared to the liver. (d) Axial image from a fused PET/CT shows intense FDG uptake in both gastric masses (arrows)
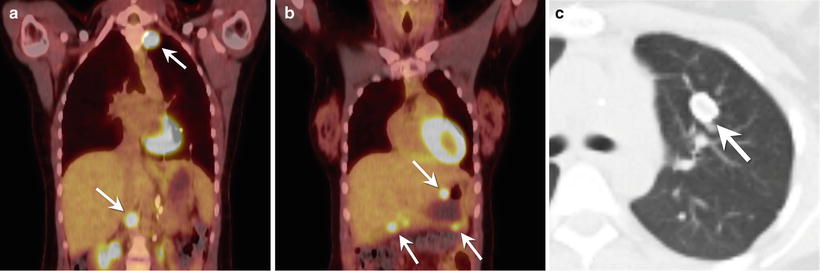
Fig. 9.5
A 17-year-old female with Carney syndrome. (a) Coronal fused image from a PET/CT through the chest and upper abdomen shows increased FDG uptake in two paragangliomas (arrows). (b) Coronal fused image from a PET/CT through the chest and upper abdomen (more anterior to the image in part (a)) shows increased FDG uptake in multiple gastrointestinal stromal tumors (arrows) in the wall of the stomach and proximal duodenum. (c) Axial CT image of the chest shows a pulmonary chondroma (arrow) in the left upper lobe. The mass has a peripheral rim of calcification
Ultrasound: On ultrasound, GIST can appear as a hypoechoic mass. Because GISTs are usually exophytic and project into the abdominal cavity, it is difficult to determine the organ of origin on ultrasound.
CT: On CT, a GIST appears as a well circumscribed mass arising from the outer wall of the gastrointestinal tract (Fig. 9.4a). Mucosal ulceration can be visible on the luminal surface of the tumor. After contrast administration (Fig. 9.4a), the mass can have variable enhancement. Calcification can also be present.
MRI: GIST has a variable appearance on MRI. It typically is isointense to the gastric wall on T1-W images and after contrast administration, the mass enhances (Fig. 9.4b). On T2-W images, GIST appears hypointense or isointense to the gastric wall (Fig. 9.4c).
Genetics
GIST can present in childhood either as a sporadic tumor or in the setting of a genetic syndrome. About 10 % of pediatric patients with GIST have a predisposition syndrome, such as Carney triad, Carney–Stratakis dyad, familial GISTs, or neurofibromatosis (NF) type 1.
The Carney triad is the most common entity predisposing pediatric patients to GIST. It is a sporadic syndrome that also includes paragangliomas and pulmonary chondromas (Fig. 9.5a–c). Carney triad GIST tends to occur in the antrum or lesser curvature of the stomach [23]. Patients with Carney triad are usually female (85 %) with a mean age at presentation of 20 years [23].
Other syndromes such as the Carney–Stratakis dyad and familial GIST syndrome are less common. The Carney–Stratakis dyad is an autosomal dominant cancer predisposition syndrome characterized by paragangliomas, GIST, and other tumors [23]. GIST in patients with Carney–Stratakis dyad tends to be multifocal and arise from the stomach. The median age at presentation is 19 years. With familial GIST syndrome, an autosomal dominant disorder, there is an activating mutation in the KIT or platelet-derived growth factor α (PDGFRA) gene [23]. Patients with familial GIST syndrome usually do not present with a tumor until they are in their mid-40s.
Pathology
Gross and microscopic features: GISTs arise from myenteric ganglion cells. They contain spindle cells, epithelioid cells, or a mixture of both (Fig. 9.6a). Pediatric GISTs are predominantly the epithelioid type [25]. Grossly they can range from small nodules to large masses.
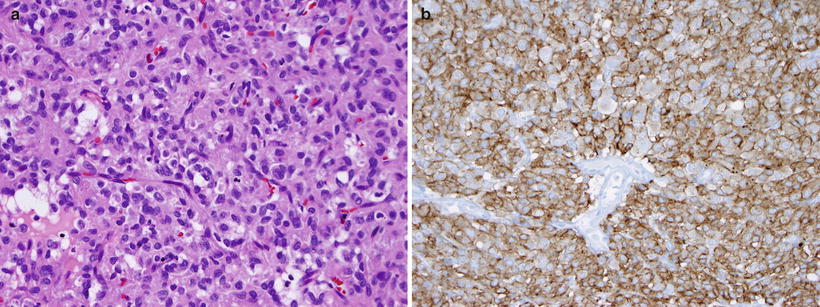
Fig. 9.6
Gastric GIST. (a) The lesion demonstrates epithelioid features and contains groups of cells with round to oval nuclei and modest amounts of lightly eosinophilic cytoplasm, separated by a rich vascular stroma. (b) The lesion strongly expresses c-kit, with membranous accentuation (CD117 immunostain)
Immunohistochemistry and other special stains: More than 95 % of GISTs express CD117 (c-Kit) on immunohistochemical stains (Fig. 9.6b), even in pediatric tumors that frequently lack mutations in this gene. DOG1 staining may be useful for patients with negative CD117 stains [26].
Molecular diagnostic features and cytogenetics: Large-scale chromosome changes have been identified in pediatric GISTs. Unlike adult tumors, KIT or PDGF mutations are infrequent. It is recommended that mutational analysis of tumor tissue be performed, as specific treatment guidelines are available for patients with KIT or PDGFR mutations. Mutations in the succinate dehydrogenase gene (SDH) are common in pediatric GISTs lacking KIT/PDGFR abnormalities [25, 27].
Prognosis and Treatment
Surgery is the mainstay of therapy for non-metastatic tumors. The goal of surgery is complete resection without violation of the tumor pseudocapsule [23]. Due to the high incidence of nodal involvement, lymph node resection is recommended. Imatinib is not usually given to pediatric patients because it lacks the efficacy seen in adults [23].
The most common prognostic system for GIST in adults utilizes tumor size, location and mitotic rate. The outcome in pediatric GIST appears more difficult to predict than that of adults. Although pediatric GIST is prone to lymph node metastasis and local recurrences, the clinical course is usually indolent [19]. In Carney triad, there may be a long time interval (up to 26 years) between the GIST diagnosis and second tumors [21].
Pancreas
Pancreatic malignancies are distinctly rare in childhood with an overall incidence of 0.018 cases per 100,000 children [28]. There were 58 cases of malignant pancreatic neoplasms in patients under 20 years of age reported to the SEER registry between 1973 and 2004 [28]. These consisted of 31 exocrine tumors (ten solid pseudopapillary neoplasm, ten pancreatoblastomas, seven ductal adenocarcinomas, and four acinar cell carcinomas), 19 NETs, and 5 sarcomas. This discussion will be restricted to solid pseudopapillary neoplasm, pancreatoblastoma, and pancreatic NET.
Solid Pseudopapillary Neoplasm (SPN)
Solid pseudopapillary neoplasm (SPN) has been called many names, including solid and papillary epithelial neoplasm, papillary-cystic neoplasm, papillary cystic tumor of the pancreas, and Frantz tumor.
Clinical Features and Epidemiology
SPN account for only 1–2 % of all pancreatic tumors, but they are the most common pediatric pancreatic neoplasms, representing over 50 % of the total [29–34]. SPNs are found almost exclusively in young women. In one large review of SPNs, more than 90 % of patients were female, and 85 % were under 30 years of age [35]. Patients with SPN typically present with nonspecific abdominal complaints and a palpable abdominal mass [36]. Other signs and symptoms can include gastrointestinal obstruction, jaundice, or pancreatitis.
Imaging Features (Figs. 9.7 and 9.8)
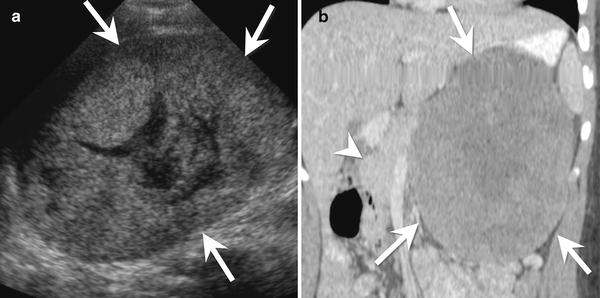
Fig. 9.7
A 15-year-old female with a solid pseudopapillary neoplasm of the pancreas. (a) Transverse ultrasound image of the upper abdomen shows a large heterogeneous mass (arrows). The mass is mostly solid with a central cystic area. (b) Coronal contrast-enhanced CT shows a large, mildly heterogeneous mass (arrows) arising from the tail of the pancreas. The mass enhances less than the remainder of the pancreas (arrowhead)
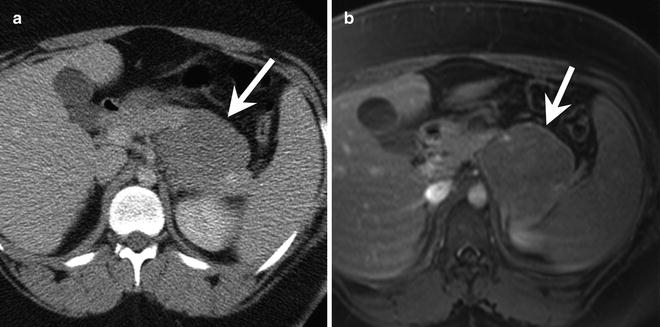
Fig. 9.8
A 15-year-old female with a solid pseudopapillary neoplasm. (a) Axial contrast-enhanced CT shows a mass (arrow) arising from the tail of the pancreas. The mass enhances less than the background pancreas. (b) Axial T1-W post-contrast MRI shows the hypoenhancing mass (arrow) arising from the tail of the pancreas
SPNs are partially cystic and partially solid masses arising from the pancreas. The tumors are usually large at diagnosis and compress adjacent structures [37].
Ultrasound: On ultrasound, a SPN appears as a large heterogeneous mass, usually well demarcated with an echogenic rim. SPNs can have variable internal characteristics. Most commonly, they have a complex internal architecture with echogenic solid components and hypoechoic cystic components (Fig. 9.7a) [37].
CT: On CT, SPNs have a hypodense rim. The solid component of the tumor is isodense to the remainder of the pancreas, while the cystic component has a density slightly greater than water [37]. Fluid-debris levels are present in up to 20 % of tumors, and calcifications can be seen in one-third of cases [37]. After the administration of contrast, SPNs enhance mildly in their solid portions. The fluid and debris do not enhance (Figs. 9.7b and 9.8a).
MRI: MR imaging shows a pancreatic mass with a hypointense capsule on T1-W and T2-W images. There is often internal high signal on T1-W images due to intratumoral hemorrhage [37]. Compared with the pancreas, the solid portions of the mass are hypointense to isointense on T1-W images and slightly hyperintense on T2-W images [37]. The enhancement pattern of SPNs on MRI resembles that seen on CT, with early peripheral, heterogeneous enhancement that gradually diffuses with time (Fig. 9.8b) [37].
Pathology
Gross and microscopic features: Grossly, SPNs form large, circumscribed tumors with degenerative and hemorrhagic change [37]. The tumors measure of 6–10 cm and occur throughout the pancreas, though some studies report a predilection for the head or tail [37]. Histologically, the tumor contains merging solid, pseudopapillary, and cystic regions (Fig. 9.9) [37]. Solid sheets of uniform, epithelial cells may resemble endocrine tumors. Characteristic perivascular pseudopapillae frequently contain pools of mucinous material. Hemorrhage and/or necrosis are common features.
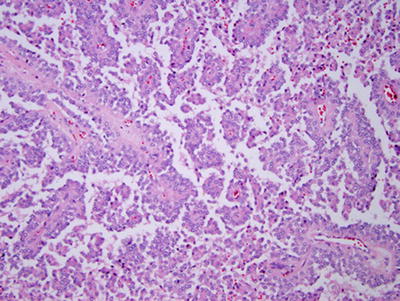
Fig. 9.9
Solid pseudopapillary neoplasm of pancreas. The neoplasm contains papillary fronds that are lined by poorly differentiated cells and appear to float in the plane of section
Immunohistochemistry and other special stains: Immunohistochemistry reveals a complex array of reactive antibodies including CD56, CD10, nuclear beta-catenin, alpha-1 antitrypsin, progesterone receptors, and synaptophysin [38, 39]. Ultrastructural studies also document acinar, ductal, and endocrine features [40].
Molecular diagnostic features and cytogenetics: Mutations of the beta-catenin gene CNNB1, nuclear accumulation of beta-catenin protein, and alterations in the Wnt-signaling pathway are commonly found in SPN [38, 41–43]. The accumulation of protein products of chromosome 11q genes, such as cyclin D1, CD56, and FLI-1, is also characteristic [44]. Allelic loss on chromosome 5q22.1 was frequently found by Min Kim et al. [42]. Between chromosomes 13 and 17 resulting in a loss of 13q14 → qter and 17p11 → pter was described in one SPN.
Prognosis and Treatment
SPNs are usually slow-growing tumors with a benign clinical course [37]. Because of the potential for aggressive behavior, they should be completely resected if possible. Nearly 85 % are confined to the pancreas, and 95 % of these are cured with resection [37]. Metastases develop in 7–16 %, usually in older women and most commonly occur in the liver [37].
Pancreatoblastoma
Clinical Features and Epidemiology
Pancreatoblastoma, the most common pancreatic tumor in young children, typically occurs in children between 1 and 8 years of age (mean age 5 years), but it also has been reported in adolescents [45]. Overall, it accounts for 0.2 % of pancreatic tumors [37] and is more common in children of Asian descent and males (M:F = 1.14:1 to 2.7:1) [37, 45]. Pancreatoblastoma has been associated with Beckwith–Wiedemann syndrome and familial adenomatous polyposis (FAP).
Patients present with a large abdominal mass, abdominal distension, and upper abdominal pain. On laboratory examination, serum α-fetoprotein (AFP) is elevated in up to 33–70 % of patients [37, 45]. Other elevated serum tests can include α-1-antitrypsin and lactate dehydrogenase. Tumor hormones can cause endocrine syndromes such as Cushing syndrome and the syndrome of inappropriate antidiuretic hormone [45].
Imaging (Fig. 9.10)
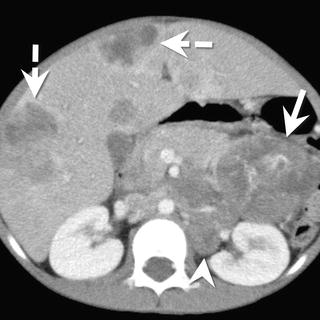
Fig. 9.10
A 6-year-old male with pancreatoblastoma. (a) Axial contrast-enhanced CT shows a large mass (arrow) arising from the pancreatic head. The mass is hypodense compared to the pancreas and contains multiple enhancing vessels. Metastatic disease is present, involving the liver (dashed arrows) and retroperitoneal lymph nodes (arrowhead)
On imaging, the mass is often very large with a mean diameter of 7–18 cm [45]. Because of the large size, it can be difficult to determine the organ or origin.
Ultrasound: At ultrasound, pancreatoblastoma appears as a heterogeneous, well-circumscribed mass with cystic and solid components [37]. The cystic structures are hypoechoic with internal septa. When solid components are present, they appear hypoechoic to the normal pancreas.
CT: On CT, pancreatoblastoma appears as a well to partially circumscribed mass arising from the pancreas [37]. The tumor has a heterogeneous appearance with hypodense cystic areas and enhancing septa. Small punctate or curvilinear calcifications may be present (Fig. 9.10) [37].
MRI: On MRI, pancreatoblastoma is hypointense to isointense on T1-W images and hyperintense on T2-W images. Due to internal necrosis, the mass has a heterogeneous appearance.
Pathology
Gross and microscopic features: Grossly, pancreatoblastomas are partially encapsulated, large (mean diameter = 10.6 cm ), solitary masses [46]. Microscopic examination shows a cellular tumor with a variety of patterns, including prominent acinar differentiation and lesser degrees of endocrine and ductal differentiation (Fig. 9.11). Often, bands of spindled stromal cells separate solid and lobular sheets of epithelial cells. Endocrine differentiation has been observed in only a few instances. Squamoid corpuscles, a characteristic feature, may range from clusters of nondescript cells to clear squamous cells with keratinization.
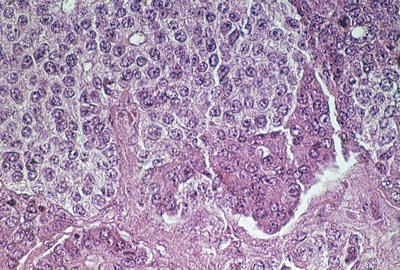
Fig. 9.11
Pancreatoblastoma. This tumor contains a mélange of primitive, eosinophilic acinar epithelium and amphophilic neuroendocrine cells
Immunohistochemistry and other special stains: Immunohistochemical stains highlight acinar differentiation with trypsin and chymotrypsin expression. Squamous corpuscles show a characteristic epithelial immunoprofile: keratins 8, 18, 19, and EMA are positive, whereas cytokeratin in negative or heterogeneously positive [47]. Immunohistochemical staining may demonstrate focal endocrine differentiation via chromogranin, synaptophysin, and neuron-specific enolase expression.
Molecular diagnostic features and cytogenetics: Genetic alterations are not well defined in pancreatoblastoma. Allelic loss of 11p has been reported [48].
Prognosis and Treatment
Surgical resection remains the mainstay of therapy for pancreatoblastoma. Primary resection may not be possible in patients with large tumors involving major blood vessels or adjacent organs. In these patients and those with metastases, neoadjuvant chemotherapy is often the first line of treatment [45]. Pancreatoblastomas have a high recurrence rate. After resection, patients are monitored with serial imaging and serum AFP levels.
Pancreatic Neuroendocrine Tumors
Clinical Features and Epidemiology
Pancreatic NETs are rare tumors in children and typically occur in middle aged adults. They may be asymptomatic or present with a hormone secretion syndrome. The latter tumors are said to be functioning or hyperfunctioning [37], whereas all other tumors are either nonfunctioning or clinically silent. Insulinomas are the most common subtype followed by gastrinomas [37].
The clinical presentation of pancreatic NET depends on tumor function. Insulinomas can present with symptoms of hypoglycemia that resolve with glucose [37]. In young children, hypoglycemia can cause behavioral problems, seizures, or coma [37]. Gastrinomas cause Zollinger-Ellison syndrome or diarrhea. Nonfunctioning pancreatic neuroendocrine tumors present later, often with symptoms due to mass effect.
Imaging (Figs. 9.12 and 9.13)
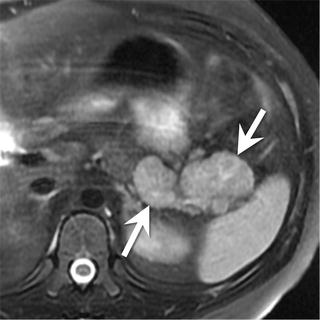
Fig. 9.12
An 11-year-old male with a pancreatic neuroendocrine tumor (secreting adrenocorticotropic hormone (ACTH)). Axial T2-W MRI of the upper abdomen shows a bilobed mass (arrows) arising from the pancreatic tail. The mass is hyperintense compared to the remainder of the pancreas
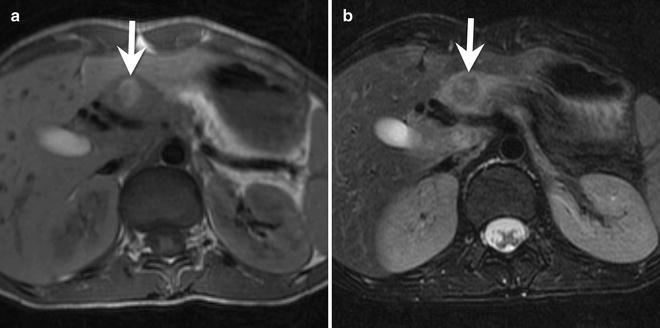
Fig. 9.13
A 15-year-old female with a poorly functioning pancreatic neuroendocrine tumor (insulinoma). (a) Axial T1-W MRI shows a hyperintense, circular mass (arrow) in the neck of the pancreas. (b) The mass (arrow) is hypointense to the pancreas on T2-W images. There was peripheral enhancement on post-contrast images (not shown). Histologically, this insulinoma has a trabecular pattern
Pancreatic neuroendocrine tumors vary in their location in the pancreas depending on the type of tumor. Insulinomas occur more commonly in the body and tail of the pancreas, whereas gastrinomas and nonfunctioning tumors are more common in the head (Fig. 9.12) [37].
Ultrasound: Insulinomas are typically small round or ovoid tumors that appear hypoechoic with a hyperechoic rim. Larger pancreatic NETs are usually heterogeneous with foci of cystic change, hemorrhage, and necrosis. There can be small echogenic areas of calcification on ultrasound [37].
CT: On CT, insulinomas are small tumors that avidly enhance during the arterial phase. The larger tumors are heterogeneous with enhancing solid portions and non-enhancing cystic areas [37]. Larger tumors are more likely to be associated with liver metastases.
MRI: Insulinomas are hyperintense on T1-W and T2-W images (Fig. 9.13). They avidly enhance on the arterial phase of imaging.
Nuclear medicine: Somatostatin receptor imaging can be useful to localize a tumor; however, only 60–75 % of pancreatic NET express somatostatin receptors.
Pathology
Gross and microscopic features: Grossly, pancreatic neuroendocrine tumors are round or ovoid tumors. Small tumors are solid and homogeneous, while larger tumors have an incomplete fibrous pseudocapsule and contain cysts, necrosis, or hemorrhage [37].
Histologically, pancreatic NETs contain sheets or nests of monomorphic cells arranged in one of three patterns: the trabecular pattern with or without a gyriform arrangement, the acinar or glandular pattern, or the medullary or solid pattern (Fig. 9.13a) [37]. The intervening stroma is variable and can contain numerous blood vessels. Insulinomas characteristically contain amyloid that shows green birefringence when stained with Congo red [37].
Immunohistochemistry and other special stains: Pancreatic NETs generally show strong reactivity with antibodies to synaptophysin and chromogranin A. Detection of one or more peptide hormones in tumor tissue is common, even in nonfunctioning tumors. Expression of cytokeratin 19 has been associated with poor outcome in several studies [49, 50] but not in others [51].
Molecular diagnostic features and cytogenetics: Familial inherited syndromes, such as the multiple endocrine neoplasia (MEN) 1, von Hippel-Lindau, NF1, or tuberous sclerosis, may be associated with pancreatic NETs. In sporadic pancreatic NET tumors, losses of chromosome 1 and 11q and gain on 9q have been identified [52].
Prognosis and Treatment
Surgical excision can cure both non-metastatic pancreatic NET and its associated clinical syndrome [37]. For tumors with metastatic disease at presentation, the optimal therapeutic approach and outcome are not clear. In one study of 19 pediatric patients with pancreatic NET, there was a 50 % 5-year survival rate [28].
Liver
The liver is the third most common site of origin for solid abdominal tumors in children, following the kidney and adrenal gland [53]. Hepatic tumors account for 0.3–2 % of all pediatric tumors [54]. This chapter discusses the more common malignant tumors hepatoblastoma, hepatocellular carcinoma (HCC), fibrolamellar HCC, and undifferentiated embryonal sarcoma (UES).
Overview of Imaging Approaches
When evaluating children with a suspected liver mass, ultrasound is often the first imaging study performed. Ultrasound has several advantages compared to other modalities, including lack of ionizing radiation, ability to provide real-time information, and performance without sedation or general anesthesia. It is used to help localize the tumor and define its internal structure. Doppler sonography can be used to determine the relationship of the tumor to adjacent vasculature, the patency of the vessels, and the presence of biliary obstruction.
CT is frequently used for additional evaluation of pediatric liver masses and has many advantages including ease of access, rapid imaging time (possibly obviating sedation), capacity for high-quality multiplanar and 3D reconstruction, ease of interpretation, and superior spatial resolution. Perhaps its greatest disadvantage is the use of ionizing radiation [55].
When evaluating liver tumors, it is helpful to determine the enhancement pattern of the mass during the hepatic arterial and portal venous phase of imaging. Because obtaining images in different phases of enhancement requires additional imaging, using CT for multiphase imaging can double the radiation dose delivered to the patient. This has led to some disagreement on the optimal CT protocol for liver tumor evaluation. Some groups propose performing multiphase imaging, while others image only in the portal venous phase of enhancement.
The concern over the radiation dose delivered by CT has led many pediatric radiologists to recommend MRI as the modality of choice to characterize liver masses. MRI’s advantages include its lack of ionizing radiation, its capacity for imaging in multiple planes and in multiple phases of contrast enhancement, and its superior contrast resolution. MRI’s biggest disadvantage is its long scan time, requiring sedation or anesthesia for young children and infants.
Recently, the hepatocyte-specific contrast agents gadoxetate disodium (Eovist/Primovist; Bayer HealthCare, Leverkusen, Germany) and gadobenate dimeglumine (Multihance; Bracco Diagnostics, Princeton, New Jersey) have been developed for MR imaging. These gadolinium-based contrast agents are bound by functioning hepatocytes and partially excreted by the biliary system [56]. Hepatocyte-specific contrast agents have proven useful in characterizing pediatric liver masses [57, 58].
Hepatoblastoma
Hepatoblastoma is an embryonal tumor arising from hepatocytes that resemble the fetal and embryonic liver [59].
Clinical Features and Epidemiology
Hepatoblastoma, the most common primary hepatic malignancy in childhood, accounts for between 65 and 80 % of all pediatric liver tumors [59–61]. It primarily occurs in young children and peaks during infancy (median age at diagnosis of 18 months) [59]. Hepatoblastoma is rare in older children; 90–95 % of new cases occur before 4 years of age [59, 62].
During the past 20 years, the incidence of hepatoblastoma has increased approximately 4 % per year, faster than for any other childhood cancer [63]. Currently, its overall incidence is 10.5 cases per million in children <1 year of age and 5.4 cases per million in children 1–4 years of age [63].
There are many known risk factors for hepatoblastoma including male gender, very low birth weight (<1,500 g), and genetic syndromes such as FAP, Beckwith–Wiedemann syndrome, hemihypertrophy, and trisomy 18 [63].
Imaging Features (Figs. 9.14, 9.15, and 9.16)
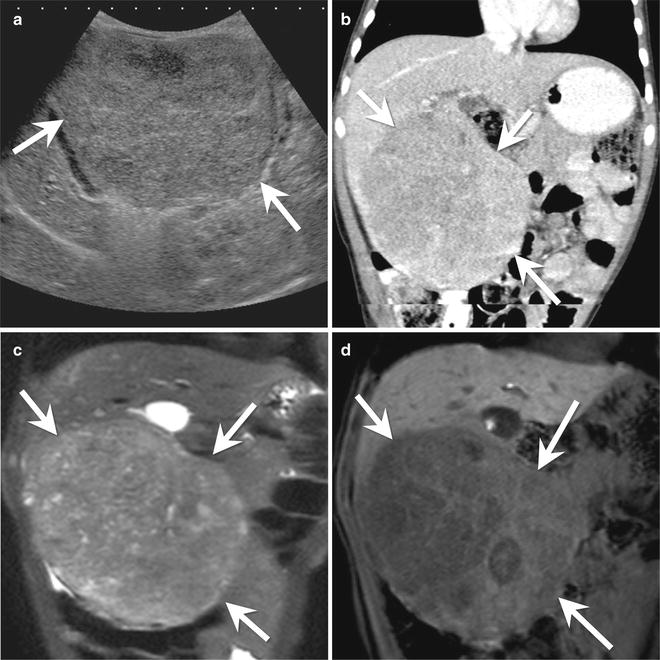
Fig. 9.14
An 18-month-old male with hepatoblastoma. (a) Longitudinal ultrasound of the liver shows a large mass (arrows) arising from the inferior aspect of the right lobe of the liver. The mass is heterogeneous with similar echogenicity to the background liver. (b) Coronal contrast-enhanced CT shows a large, pedunculated mass (arrows) arising from segments 5 and 6 of the liver. The mass is overall hypodense compared to the background liver. (c) Coronal T2-W image of the liver shows the heterogeneous appearance of the mass (arrows). (d) Coronal Liver Acquisition with Volume Acquisition (LAVA) image of the liver in the hepatocyte phase after contrast administration shows minimal enhancement of septations within the mass (arrows). The background liver is bright due to retention of the hepatocyte specific contrast agent
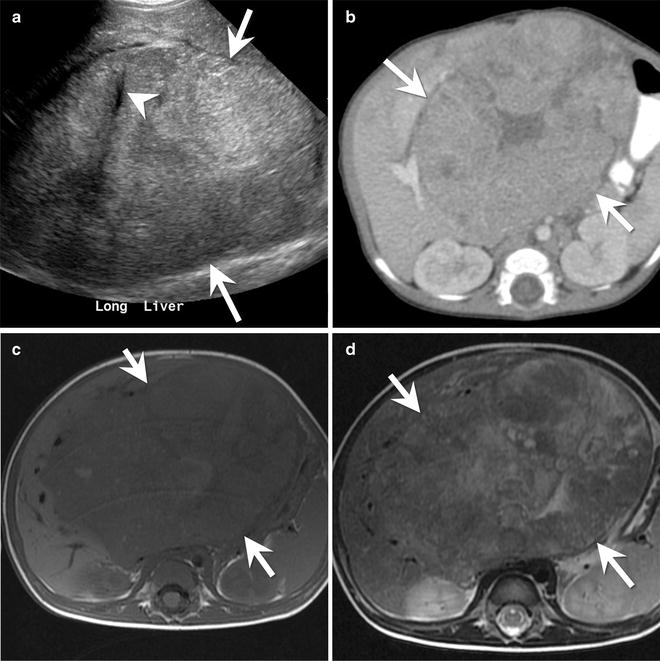
Fig. 9.15
A 4-month-old male with a mixed epithelial hepatoblastoma. (a) Longitudinal ultrasound image of the liver shows a large heterogeneous mass (arrows) arising from the liver. The mass has small areas of calcification with posterior acoustic shadowing (arrowhead). (b) Axial contrast-enhanced CT of the abdomen shows the large mass (arrows) arising from the left lobe of the liver. The mass has a central stellate area of low density. (c) Axial T1-W and (d) axial T2-W images of the liver show the large heterogeneous mass (arrows). The mass is hypointense to the background liver on the T1-W image and slightly hyperintense to the liver on the T2-W image
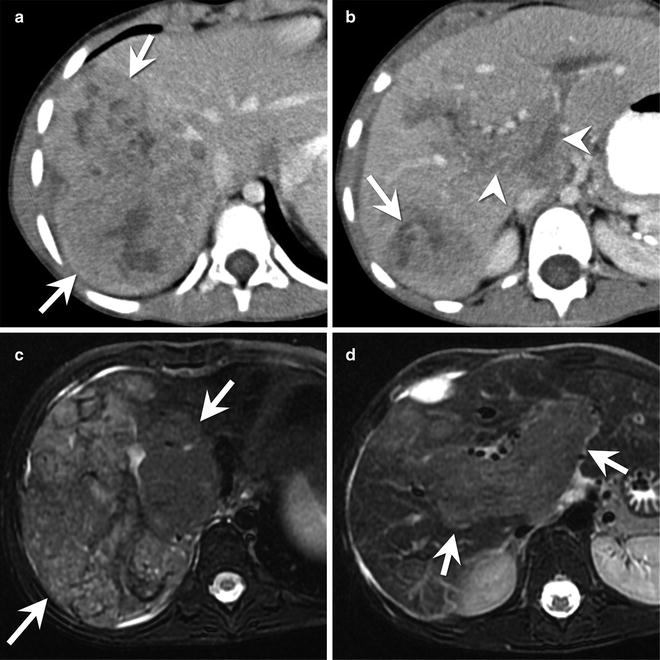
Fig. 9.16
A 6-year-old female with a mixed epithelial hepatoblastoma. (a) Axial contrast-enhanced CT shows a heterogeneous mass (arrow) within segment 7 of the liver. The mass has an irregular contour with capsular retraction along its lateral margin. (b) Axial contrast-enhanced CT image several centimeters inferior to the mass shown in part (a) shows enlargement and enhancement of the main portal vein as well as its major branches (arrowheads) consistent with tumor thrombus. A portion of the main tumor mass (arrow) is present in the posterior right lobe. (c) Axial T2-W MR shows a large, heterogeneous mass in the posterior section of the right hepatic lobe. (d) Axial T2-W MR image several centimeters inferior to the mass shown in part (c) shows enlargement and abnormal signal within the portal vein and its major branches (arrows) consistent with tumor thrombus
Imaging is used for diagnosis and staging of hepatoblastoma. The combination of the clinical and imaging findings in a child between 6 months and 3 years of age is essentially diagnostic of hepatoblastoma [65]. Hepatoblastoma can present with a solitary mass, a dominant mass with satellite lesions, or as multifocal disease [65].
Radiography: Abdominal radiography shows hepatomegaly and displacement of loops of bowel. Occasionally calcifications can be identified in the right upper quadrant [64].
Ultrasound: Ultrasound is often used as the first imaging test in a patient with hepatoblastoma. It can confirm the presence of a mass, localize the mass to the liver, and evaluate its vascular supply [65]. The tumor is usually hyperechoic compared to the background liver and shows a heterogeneous echotexture (Figs. 9.14a and 9.15a) [65]. Calcifications appear as hyperechoic foci with posterior acoustic shadowing (Fig. 9.15a). Necrosis and internal hemorrhage appear as anechoic intratumoral foci [65]. Color Doppler interrogation assists in evaluating the patency of the hepatic veins, inferior vena cava, and portal veins.
CT: Generally, hepatoblastoma appears as a well-defined, hypodense mass of the liver (Fig. 9.14b, c; Figs. 9.15b and Fig. 9.16a, b) [64]. Calcifications are present in approximately 50 % of tumors [64]. The calcifications are usually small and fine in epithelial tumors and coarse in mixed mesenchymal-epithelial tumors [65].
After the administration of contrast, the tumor enhances to a lesser degree than the background liver [64, 65]. During the arterial phase of enhancement, there may be septa or a peripheral rim of enhancement [64, 65].
Metastases are present in 20 % of patients at diagnosis and almost always occur in the lungs. Because of this, a chest CT should be performed at diagnosis [65].
MRI: The MRI appearance of hepatoblastoma varies with histologic subtype. Epithelial tumors have a homogeneous appearance and are hypointense on T1-W images (Fig. 9.15c) and hyperintense on T2-W imaging (Figs. 9.15d and 9.16c) [65]. Mixed epithelial-mesenchymal tumors are heterogeneous due to hemorrhage, necrosis, fibrosis, calcification, cartilage, and septa [65]. The variable composition of these tumors imparts a heterogeneous appearance on T1 and T2-W images. Septa appear hypointense on both T1 and T2-W images.
Post-contrast images are performed in the hepatic arterial and portal venous phases. The tumor enhances to a lesser degree than the background liver. Post-contrast imaging also defines the vascular anatomy and determines the presence of vascular invasion. Tumor thrombosis appears as an enhancing mass enlarging the involved vessel (Fig. 9.16d). This differs from bland thrombosis, which neither enhances nor enlarges vessels.
With hepatocyte-specific contrast agents, hepatoblastomas [57, 58] can have a variable appearance during the hepatocyte phase of imaging (Fig. 9.14d). Most tumors are hypointense during the hepatocyte phase, but a small number variably retain contrast [58]. It is not yet known if contrast retention occurs more commonly in certain histologic subtypes.
Pathology
Gross and microscopic features: Grossly, hepatoblastoma usually appears as a well-defined liver mass. About 60 % occur in the right lobe [66]. The tumor may have a variegated appearance due to its mixed histologic composition and areas of hemorrhage, necrosis, or calcification [64].
Histologically, hepatoblastoma is classified into two broad types with multiple distinct patterns (Table 9.1) (Figs. 9.17, 9.18, 9.19, and 9.20) [64]. The epithelial type of hepatoblastoma occurs most commonly and comprises multiple subtypes including fetal, embryonal, small cell undifferentiated, and rhabdoid (Fig. 9.17) [60]. Nearly one-third of hepatoblastomas are well differentiated epithelial tumors with pure fetal histology (Fig. 9.18) [64]. These tumors are composed of small cuboidal cells arranged in thin trabeculae and containing varying amounts of glycogen and lipid. Like fetal liver, these characteristics impart an alternating light–dark pattern at low-power microscopy. More poorly differentiated epithelial tumors contain both fetal and embryonal components (Fig. 9.17). Embryonal tumors contain small, round, blue cells with less cytoplasm than fetal cells, often arranged in epithelial rosettes. The most poorly differentiated epithelial hepatoblastoma, the small cell undifferentiated tumor (Fig. 9.19), totally lacks recognizable hepatocytic differentiation and fails to express INI1 [64]. At times, these lesions exhibit a rhabdoid phenotype, with cytoplasmic inclusions and positivity for epithelial and mesenchymal markers (see Chap. xx, Renal for further discussion).
Table 9.1




Histological subtypes of hepatoblastoma [Data from Cynthia E. Herzog, Richard J. Andrassy, Farzin Eftekhari: Childhood Cancers: Hepatoblastoma, The Oncologist 2000; 5:445–453]
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
