GENETIC CAUSES OF HEARING LOSS: SYNDROMIC AND NONSYNDROMIC
KEY POINTS
- Computed tomography and magnetic resonance imaging are critical to medical decision making in developmental abnormalities of this region and can help anticipate syndromic relationships of this as well as other organ systems.
- Reports must be logically constructed and comprehensive, anticipating all anatomic and pathologic findings that might lead to altered treatment plans.
Deafness due to a genetic defect may be syndromic or nonsyndromic. Most cases of hereditary deafness are classified as nonsyndromic, and the inner ear involvement is an isolated finding.1 Syndromic hearing loss may be conductive, sensorineural, or mixed. The functional loss may be bilateral or unilateral, and it is often asymmetric. Hundreds of forms of syndromic deafness have been identified. Clues to the presence of a syndrome include cervical and facial dysmorphism, visual system anomalies, endocrine dysfunction, cardiac problems, renal abnormalities, and hair and skin changes.1 Genomic anomalies have been established for many syndromic and nonsyndromic disorders. Some of the more common syndromic forms of hearing loss are discussed in the following material.
Genetic associations may help to anticipate and limit the damage of possibly associated genetic errors and in the future perhaps lead to some preventive strategies. Such information already facilitates genetic counseling. Recognizing the common pathways of genomic errors can also lead to prompt evaluation of other organ systems dependent for normal development on that part of the genetic code and perhaps limit morbidity due to those errors. For instance, since ear malformations are associated with an increased frequency of renal anomalies, renal ultrasound is often obtained if a syndromic etiology is suspected. Such recognition may also yield a diagnosis that had not been suspected previously. Examples include cochlear anomalies seen as characteristic of the branchiootorenal syndrome and the combination of ocular, nasal, and inner ear findings of CHARGE syndrome (Figs. 105.17 and 107.1).
REPORTING RESPONSIBILITIES AND WHAT THE TREATING PHYSICIAN NEEDS TO KNOW
The written report of findings in anomalies of the temporal bone should reflect the developmental background of these anomalies. Such a report must include systematic documentation of the entire auditory pathway as seen on either computed tomography (CT) or magnetic resonance imaging (MRI), and a specific statement for each structure in this linkage should appear in every report. A reasonable approach to the general report format is to trace the sound, essentially vibrating air, from the outside to the brain stem and the facial nerve back out. For the purposes of discussion, the suggested report content in patients with syndromic hearing loss can be relatively conveniently divided into six basic categories:
- Those related to external auditory canal atresia/middle ear anomalies (Chapter 105)
- Those related to possible inner ear anomalies (Chapter 106)
- Those that might impact cochlear implant candidates (Chapter 136) or those who are temporal bone surgical candidates for other reasons
- Anatomy of the facial nerve
- Visible and possibly related syndromic findings or clearly identifiable syndromes
- Suggestions for imaging or other clinical evaluation of organ systems that might express genetic defects
For the external canal, middle ear, and facial nerve, the report should emphasize the conductive part of the pathway but must also consider the sensorineural pathway. Facial nerve development is at the interface of these two functional units of hearing, and consideration of its anomalous development is most linked to disorders of the external canal and middle ear, where it can dramatically affect treatment options. For the patient with sensorineural hearing loss (SNHL), the report should emphasize the sensorineural part of the pathway but must also consider the preceding conductive pathway. The facial nerve position must also be considered in cochlear implant planning.
For the conductive pathways:
- Exact derangements of the conductive hearing pathway reported in a logical manner
- Status of the stapes
- Exact position of the facial nerve in the middle ear and in relationship to the atretic plate after the nerve passes caudal to the stapes
- Presence of distorted anatomy that might create problems at surgery, including the status of the mastoid and middle ear roof, position of major vessels, and any vessel that may travel an anomalous path through the temporal bone
- Whether there is an acquired or congenital cholesteatoma
- If there is an associated anomaly of the inner ear
For the sensorineural pathways:
- Exact derangements of the sensorineural hearing pathway reported in a logical manner, especially the status of the cochlea and cochlear nerve
- If there is an associated anomaly of the external or middle ear
- If there are any other associated abnormalities to suggest that this is a syndromic situation
- Whether there is any potential for cerebrospinal fluid leakage, a potential for perilymphatic fistula/gusher, or a potential route for meningitis
- Whether there are alternative or additional findings that would explain the SNHL
For both:
- Presence of distorted anatomy that might create problems at surgery, including the status of the mastoid and middle ear roof, position of major vessels, and any vessel that may travel an anomalous path through the temporal bone
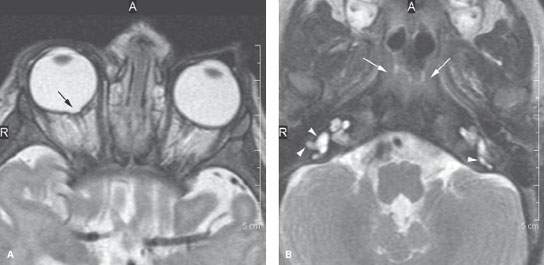
FIGURE 107.1. Findings associated with CHARGE. Magnetic resonance imaging shows coloboma (arrow in A) and suggests in (B) the presence of choanal atresia (arrows). It also shows an anomalous appearance of the vestibules on both sides (arrowheads).
EMBRYOLOGY
The various degrees of aural dysplasia and dysgenesis of the cochlea and vestibular system observed with imaging are growth errors and arrests of the branchial apparatus and otocyst projections at various stages of development. At the extreme are severe dysplasia and agenesis of structures in the conductive and sensorineural pathways. At the other end of the spectrum from an imaging perspective are mild dysplasias or just physiologic abnormalities limited to the membranous labyrinth that will have no, or only indirect, abnormal imaging evidence of their presence. The latter may in the future be more appropriately anticipated by genetic markers. Some growth arrests can be reasonably linked to the embryologic stage of development and further associated with specific genetic errors, in some cases syndromic. Those syndromes are the main topic of this chapter. Those errors in development are discussed more specifically in Chapters 105 and 106.
SPECIFIC GENETIC SYNDROMES AND RELATED IMAGING FINDINGS
Oculoauriculovertebral Spectrum
The oculoauriculovertebral spectrum (OAVS) is a group of disorders that have unilateral or bilateral abnormalities of the first and second branchial arches. These errors result in facial asymmetry and mandibular and/or maxillary hypoplasia.
OAVS includes hemifacial microsomia (HFM), Goldenhar syndrome (GS), the first and second branchial arch syndrome, and facioauriculovertebral syndrome.
Expressions of OAVS errors are generally associated with a spectrum of aural atresia and sometimes ocular abnormalities. GS has bilateral facial microsomia, vertebral anomalies and epibulbar dermoids, and sometimes abnormalities of the other organ systems (Figs. 107.2 and 107.3). CT of the temporal bones in HFM and GS reveals unilateral or bilateral stenosis or atresia of the external auditory canal, a small middle ear cavity, ossicular malformation, and atresia or stenosis of the oval window. Inner ear anomalies also sometimes occur in patients with GS.
Otocraniofacial or Craniosynostosis Syndromes
Crouzon disease (otocraniofacial dysostosis) manifests with outer and middle ear malformations, such as microtia and external auditory canal stenosis or atresia and a narrow middle ear cavity with ossicular malformation and stenosis of the round window. The inner ear is otherwise not affected. The facial abnormalities are characterized by hypoplasia of the maxilla, shallow orbits, proptosis, and craniosynostosis.
Apert syndrome or acrocephalosyndactyly is frequently associated with deafness. Hearing loss is usually conductive due to stapes fixation. Inner ear abnormalities are poorly characterized. These children have bicoronal synostosis, midface hypoplasia, and complex syndactyly of the hands and feet. The most frequently associated cerebral malformations are hypoplasia of the corpus callosum and septum pellucidum and ventricular enlargement. Optic nerve atrophy is frequently seen.
Robin Sequence
The Robin sequence is characterized by cleft palate, glossoptosis secondary to micrognathia both of which produce feeding difficulties. The Robin sequence can be sporadic or syndromic. The most common are Stickler and velocardiofacial syndromes. The hearing loss is variable and most often conductive, especially in younger children. It may be due to eustachian tube dysfunction and middle ear effusion. Ear malformations include deformed, low-set ears; abnormal stapes; a small or dehiscent facial nerve; anomalies of the cochlea and semicircular canals; and small internal auditory canals.
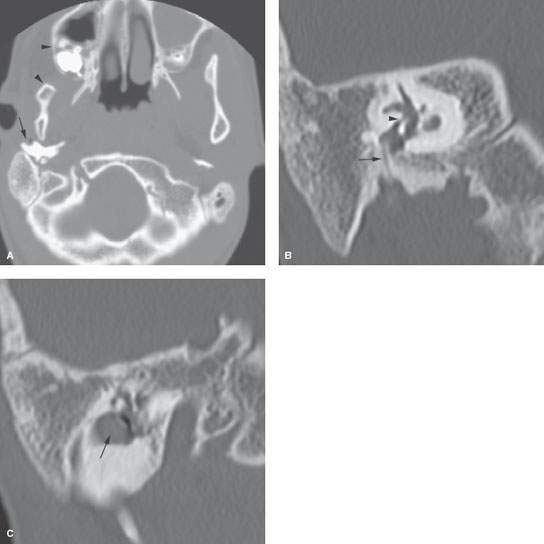
FIGURE 107.2. A patient with Goldenhar syndrome. A: Right side of the face microsomia manifesting as a small maxilla and mandible (arrowheads). There is also a dysmorphic tympanic bone (arrow). B: Coronal computed tomography of the temporal bone showing aural atresia and an irreparable middle ear with the facial nerve (arrow) descending through a thick atretic plate and markedly contracted oval window niche with no normal stapes present (arrowhead). C: A coronal section more anteriorly showing an associated congenital cholesteatoma that needed to be followed with imaging to determine the eventual need for removal.
Related posts:
 Fibrous Dysplasia and other Fibro-osseous Lesions of the Craniofacial Skeleton
Fibrous Dysplasia and other Fibro-osseous Lesions of the Craniofacial Skeleton
 Intraconal Orbit: Tumors
Intraconal Orbit: Tumors
 Hypopharynx: Developmental Abnormalities
Hypopharynx: Developmental Abnormalities
 Tumors of the Submandibular Gland and Space and Tumorlike Conditions
Tumors of the Submandibular Gland and Space and Tumorlike Conditions
 Masticator Space, Buccal Space, and Infratemporal Fossa Infections and Other Inflammatory Conditions
Masticator Space, Buccal Space, and Infratemporal Fossa Infections and Other Inflammatory Conditions
 Oral Cavity and Floor of the Mouth: Malignant Tumors
Oral Cavity and Floor of the Mouth: Malignant Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


