Neurofibromatosis Type 1
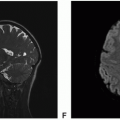
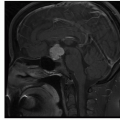
Definition: Neurofibromatosis (NF) type 1 is one of the most common inherited disorders and the most common neurocutaneous syndrome. NF1, also known as von Recklinghausen disease, is an autosomal dominant disorder due to genetic mutations of the NF1 gene on chromosome 17q11.2 and characterized by neurofibromas, multiple café-au-lait spots, axillary and inguinal freckling, optic gliomas, osseous lesions, and iris hamartomas. Slightly increased risk for malignant nerve sheath tumor, gastrointestinal stromal tumor, rhabdomyosarcoma, juvenile chronic myeloid leukemia, duodenal carcinoids, medullary thyroid carcinomas, and pheochromocytoma has been noticed.
Epidemiology: Prevalence of NF1 is approximately 1 in 3,000-3,500 births, regardless of ethnic and racial background, and about 50% of the cases are familial; the remaining cases are de novo mutations occurring primarily in paternally derived chromosomes.
Molecular and genetic profile: Mutational inactivation of the NF1 gene locus on chromosome 19q11.2 can be seen.
Clinical features and standard therapy: Multiple sites and organ systems may be involved in NF1, with the central and peripheral nervous system, the skin, the eyes, and the bones being the most common sites. Café-au-lait spots are generally the heralding feature of NF1 and bring the patient for medical examination. The first step in management of patients with NF1 is genetic counseling. It is essential to examine parents to determine whether they are affected. Regular medical appointments to a multidisciplinary clinic with annual examinations are necessary. Imaging studies are indicated only when patients are symptomatic. Surgery is currently the only treatment option for most NF1 lesions, and chemotherapy is helpful with optic pathway gliomas and higher grade astrocytomas. No effective chemotherapy is available to treat plexiform neurofibromas.
Neurofibromatosis Type 2
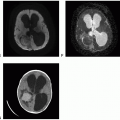
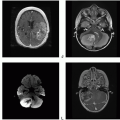
Definition: Neurofibromatosis type 2 is a rare, autosomal dominant disorder caused by mutations of NF2 gene on chromosome 22q12 and characterized by noncancerous neoplastic and dysplastic lesions that primarily affect the nervous system, with bilateral vestibular schwannomas as a diagnostic hallmark. Schwannomas can occur involving other cranial nerves, spinal and peripheral nerves, intracranial and spinal meningiomas and ependymomas, and a variety of nonneoplastic and dysplastic/hamartomatous lesions such as meningioangiomatosis, glial hamartomas, ocular abnormalities, and neuropathy.
Epidemiology: Prevalence is around 1 in 60,000.
Molecular and genetic profile: NF2 is inherited as an autosomal dominant trait in some patients or de novo mutation in the NF2 gene, which is located in the long arm of chromosome number 22 (22q12.2). The NF2 gene encodes for the protein known as merlin, which is found in the Schwann cells in the nervous system and acts as a tumor suppressor gene.
Clinical features and standard therapy: Signs and symptoms usually appear during adolescence or early in the second decade, with hearing loss, tinnitus, and balance problems being the most common. For those patients with bilateral vestibular schwannomas, a major goal of treatment is to preserve the hearing for as long as possible. Surgery is usually done in patients who have lost hearing, and radiation therapy can be done for any residual tumor. Recently, bevacizumab (Avastin), a humanized antivascular endothelial growth factor monoclonal antibody that interferes with blood vessel formation, has been shown to slow the growth of vestibular schwannomas and to partially restore hearing in some patients and holds promise that other drugs may be proven effective as well.










 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access