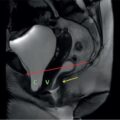
Priscilla Joshi, Vandana Jahanvi Renal anomalies encompass a spectrum which ranges from the lethal renal agenesis to incidentally detected renal anomalies secondary to aberrant embryonic migration. Imaging helps in the early diagnosis, detection of complications, surgical planning and follow-up of these patients. The kidneys develop from the pronephros, mesonephric and metanephros. These succeed each other. The intermediate mesoderm develops into the pronephros at the end of the 3rd week of gestation. This regresses a week later and is replaced by the mesonephros. The kidneys develop during the 4th week of gestation. The ureteric buds fuse with the metanephros. This occurs at the level of the first two sacral segments. In the pelvis, the kidneys are close to each other with the hila directed anteriorly. Over the next 4 weeks, the kidneys ascend into the lumbar region and move away from each other as well as rotate medially so that the hila are medially and anteriorly directed. Congenital urinary tract anomalies range from anomalies in number, anomalies in shape, cystic renal disease, collecting system anomalies and urinary bladder anomalies. These will be discussed one by one in this section. In renal hypoplasia the kidneys are small. They show a normal architecture, but the number of nephrons is reduced. The condition can be unilateral or bilateral. If unilateral the contralateral kidney shows compensatory hypertrophy. Hypoplasia if bilateral is part of genetic syndromes like renal coloboma syndrome and branchio-oto-renal syndrome. It can result in end-stage renal disease because of the reduced number of nephrons. US: A kidney which is smaller by 2 Sd as compared to the mean kidney size expected at that age. Radionuclide scan (DMSA): Rules out scarring. Less than 100 cases of this anomaly have been reported in literature. This is an extremely rare anomaly. The accessory kidneys are usually seen on the left, caudal to the left kidney. Accessory kidneys are usually smaller in size and show suboptimal function. They can be detected on ultrasound and confirmed with cross-sectional modalities like MR urography/CT urography. Function can be evaluated on postcontrast CT/MR urography or with dimercaptosuccinic acid (DMSA) and diethylene triamine penta-acetic acid (DTPA) scans. Associated urogenital or other anomalies maybe be seen. Incomplete fusion of the developing renal lobules causes a lobulated appearance of the kidneys. This has to be differentiated from renal scarring. Persistent foetal lobulations are seen as a smooth indentation of the renal outline. This is seen between the renal pyramids on ultrasound whereas the indentation in scarring overlies the renal pyramids and is not smooth and symmetrical. This can be appreciated on US, CT and MRI. This occurs due to prominent renal cortical tissue between the pyramids extending towards the parenchyma. It can mimic a renal mass on ultrasound. The interpolar region of the left kidney is a common site. It can be differentiated from a mass as it is in continuity with normal renal parenchyma and shows the same appearance as the rest of the renal parenchyma on all imaging modalities. This is a normal variation in the contour of the left kidney. The lower pole of the spleen causes an impression on the superolateral aspect of the kidney, which can mimic a renal mass. It shows the same echogenicity/density and signal intensity as the rest of the kidney. Postcontrast images on CT and MR also show enhancement akin to the rest of the kidney. Also, the calyces extend further laterally into the hump as compared to the other calyces. Infolding of the cortex at the level of the renal sinus can appear as a pseudomass, known as the renal hilar lip. As in the case of the dromedary hump the imaging characteristics of this lesion being akin to the rest of the renal tissue help reaching a diagnosis. This occurs due to embryonic fusion of renunculi. It is a normal variant which is located in the interpolar region. It occurs due to extension of sinus fat into the cortex and is seen as a triangular echogenic area on ultrasound which is in contiguity with the renal sinus fat. Differential diagnosis can include a renal angiolipoma which on ultrasound is well defined, echogenic, round, and is not in contiguity with the renal sinus fat. Simple renal cysts are rarely seen in children. An underlying genetic cystic disease or rarely a malignancy needs exclusion. The diagnosis of a simple cyst should be one of exclusion. At least one follow-up ultrasound should be done to evaluate change in size and imaging appearance as well as to rule out development of additional cysts. Further cross-sectional imaging evaluation with CT and MRI and contrast studies are not required unless atypical findings are seen initially or on follow-up. This is a severe form of renal dysplasia which may be unilateral or bilateral. Renal dysplasia occurs due to abnormal metanephric differentiation. It has an incidence of 0.3 to 1 in 1000 live births. When bilateral it is incompatible with life. The kidney is not reniform and is composed of multiple cysts of varying sizes which are noncommunicating. There is no visible functioning renal parenchyma appreciable. The collecting system and ureter are both not seen/atretic. The condition is now being detected more often on antenatal ultrasound foetal MRI (Fig. 10.7.1). The contralateral kidney, if normal, shows compensatory hypertrophy. Sequelae such as hypertension, proteinuria and renal impairment are uncommon as is contralateral VUR, PUJO. There is no increased incidence of malignancy reported. US: Multiple cysts of varying sizes which are noncommunicating are seen. The kidney may be enlarged and even palpable in neonates. Subsequently, the kidney may involute. The contralateral kidney shows compensatory hypertrophy. CT/MR: It shows similar findings. The kidney is nonfunctioning on postcontrast CT/MR or a DMSA scan. The ureter may be atretic. ARPKD is a paediatric cystic renal disease with an incidence of 1 in 10,000 to 40,000 live births. There is no gender predilection. This condition results from mutations in the PKHD1 gene on chromosome 6p12 that encodes for the protein fibrocystin. The earlier the diagnosis is made the condition will be more severe. This condition manifests itself at the age of 30–40 years. It is one of the most common genetic disorders caused by a single gene mutation with an incidence of 1 in every 1000 individuals The kidneys are enlarged (Fig. 10.7.4) and contain multiple cysts of varying sizes. There is deterioration of renal function leading to renal insufficiency. In the neonate, the cysts may be very small and discrete. In children without a genetic diagnosis or a clear family history where the condition is detected, a follow-up USG should be performed within 12 months of the initial detection.
10.7: Genitourinary anomalies
Introduction
Embryology
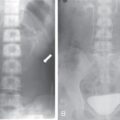
Renal hypoplasia
Imaging
Anomalies in number
Supernumerary kidneys
Anomalies in shape
Persistent foetal lobulations
Hypertrophied column of bertini
Dromedary hump
Renal hilar lip
Junctional parenchymal defect
Cystic renal diseases
Simple cyst
Multicystic dysplastic kidney
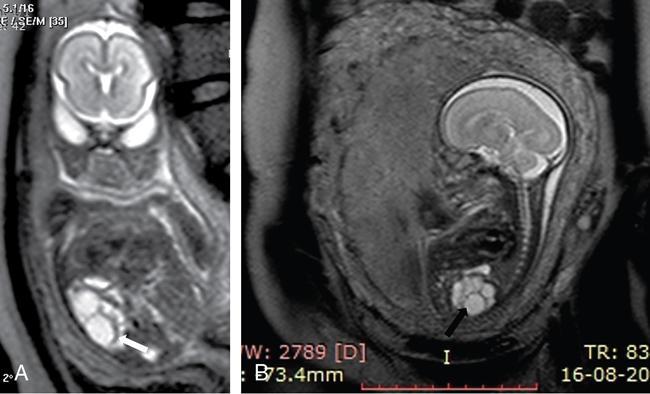
Imaging findings (Fig. 10.7.2)
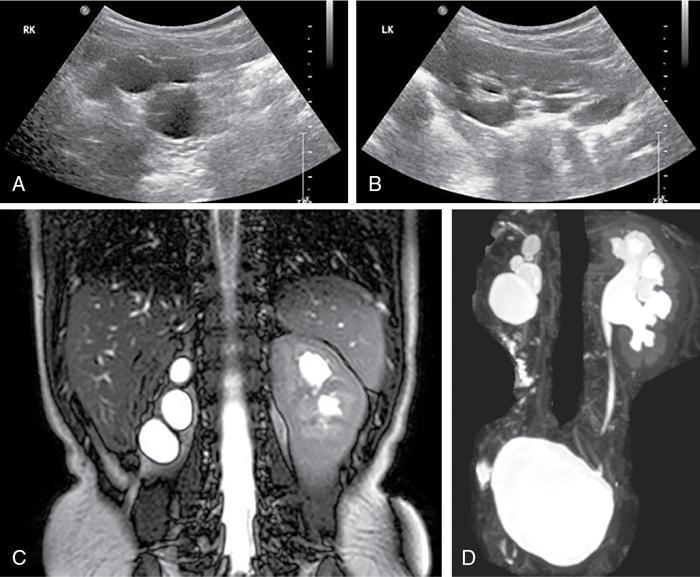
Autosomal recessive polycystic kidney disease (ARPKD)
Imaging findings (Fig. 10.7.3)
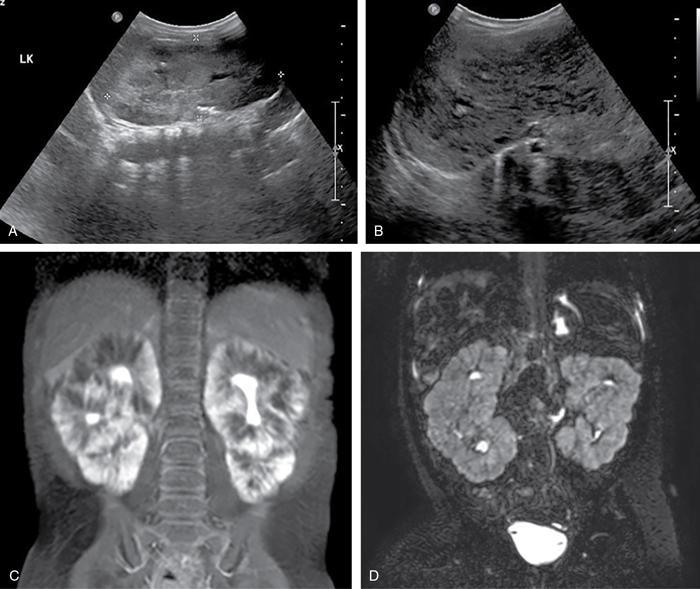
ADPKD: Autosomal dominant polycystic kidney disease
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Radiology Key
Fastest Radiology Insight Engine