(1)
Department of Clinical Radiology, Amiri Hospital – Kuwait City, Kuwait City, Kuwait
9.3.1 The Lungs in SCD
9.3.3 The Brain in SCD
9.3.4 The Spleen in SCD
9.5 Hemophilia
9.6 Lymphomas
9.7 Leukemia
9.9 Amyloidosis
9.10 Evans’ Syndrome
9.11.3 Kimura’s Disease
9.12 Mastocytosis
9.1 Hemosiderosis and Hemochromatosis
Hemosiderosis, or iron overload, is a pathological condition characterized by deposition of excess iron within the body tissues that normally do not contain iron. Hemosiderosis is usually secondary to a primary cause such as multiple blood transfusion, chronic hemodialysis, or hemolytic anemia (e.g., thalassemia).
When iron is released into the cytoplasm, it enters a cellular compartment called “labile iron pool” (LIP). Both ferrous (Fe2+) and ferric (Fe3+) iron forms are poorly bound with proteins and are highly toxic to the cells within this compartment. The unneeded iron from LIP is stored in the form of ferritin, which is organic and nontoxic. When the LIP iron content exceeds the ferritin capacity, hemosiderin is generated from ferritin denaturation. Iron in the form of hemosiderin is thought to be more toxic to the body tissues. Hemosiderin initially accumulates in the reticuloendothelial system (spleen, bone marrow, and Kupffer cells in the liver). When the reticuloendothelial system is saturated, deposition occurs in normal body tissues such as the hepatocytes, heart muscles, and the endocrine system. Chelation therapy (e.g., desferrioxamine) removes mainly the extracellular iron and only a fraction of the intracellular LIP iron.
Hemosideroses have different body manifestations according to the area of deposition:
Cardiac hemosiderosis: myocardial iron deposition results in dilated cardiomyopathy that will lead to heart failure.
Signs on Chest Radiograph
The heart appears larger than normal in cases of cardiac damage and development of dilated cardiomyopathy.
Signs on MRI
MRI can detect early myocardial siderosis via obtaining T2* images, which show a dark, hypointense rim located within the myocardium in short-axis sequences, representing myocardial siderosis (Fig. 9.1.1).

Fig. 9.1.1
Short-axis cardiac MRI illustration demonstrates a black ring within the myocardium, a sign of cardiac siderosis (arrowhead)
Hepatic hemosiderosis: iron deposition in hepatocytes results in liver cirrhosis.
Signs on CT and MRI
On CT, the liver appears hyperdense compared to the spleen.
On MRI, the liver appears extremely hypointense, with an almost black signal on all pulse sequences, depending on the severity of the iron overload.
Anterior pituitary gland hemosiderosis: loss of the endocrine function of the anterior pituitary results in hypogonadism and loss of libido.
Signs on MRI
The anterior pituitary shows a hypointense signal intensity area on both T1 and T2 images.
Pancreatic hemosiderosis: deposition of iron in the pancreases results in impairment of both exocrine and endocrine functions. Diabetes mellitus may result due to pancreatic siderosis.
Signs on US
The normal pancreas has almost the same echogenicity as the liver on ultrasound. In siderosis, the pancreas may appear hyperechoic compared to the liver, due to iron overload. Other causes of hyperechoic pancreas include fatty infiltration and pancreatic calcification.
Signs on MRI
Pancreatic hemosiderosis are detected as hypointense signal intensity areas are seen within the pancreas on both T1W and T2W images.
Hemochromatosis is a disease characterized by deposition of excess iron in the body as a result of genetic defect (primary hemosiderosis).
In hemochromatosis, iron deposition initially occurs in the hepatocytes and spares Kupffer cells (the reverse situation to hemosiderosis). The age of presentation is usually between 50 and 60 years of age. Menstruation blood loss in women has a protective effect against hemochromatosis due to iron loss.
Hemochromatosis is asymptomatic in early stages. As the iron deposition progresses, liver failure, skin hyperpigmentation, arthropathy (especially in the metacarpophalangeal joints), and cardiac failure may occur. Deposition of hemosiderin in the subcutaneous tissues results in increased skin tanning and darkening.
Bronze diabetes is a term used to describe maturity-onset diabetes seen in hemochromatosis. The term “bronze” is used because the diabetes is associated with skin tanning that mimics bronze coloring.
Diagnosis is confirmed by measuring serum ferritin level, transferrin saturation testing, liver biopsy, genetic testing, and MRI.
Differential Diagnoses and Related Diseases
Bantu siderosis a type of hemosiderosis only found in Africa. It is associated with liver cirrhosis, diabetes, and heart disease. The disease is linked with higher rates of tuberculosis infection.
Juvenile hemochromatosis is a form of hemochromatosis present in the second decade of life. Patients often present with abdominal pain, cardiac arrhythmias, impaired glucose tolerance, and hypogonadotropic hypogonadism.
Ferroportin disease is a genetic disease characterized by mutation in the gene responsible for the production of ferroportin, a protein that exports iron from body cells. Ferroportin is expressed mainly in Kupffer cells and splenic macrophages. Patients present with isolated hyperferritinemia and normal or slightly elevated transferrin saturation. In contrast to hemochromatosis, iron is mainly deposited within Kupffer cells.
Pulmonary hemosiderosis is a rare condition that arises due to repeated episodes of bleeding within the lung alveoli. Iron overload within the lungs results in pulmonary fibrosis, anemia, and (rarely) death due to pulmonary hemorrhage. The disease has an incidence of less than 1:1,000,000 live births and occurs usually in children <7 years old (it is extremely rare in adults). Symptoms include coughing blood (hemoptysis) and iron-deficiency anemia.
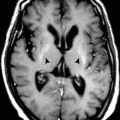
Superficial brain siderosis is a condition characterized by deposition of hemosiderin within brain tissues, often secondary to subdural hemorrhage and bleeding into the brain cisterns. When siderosis affects the vestibulocochlear nerve, tinnitus may result.
Signs on MRI
The liver appears extremely hypointense on all pulse sequences, depending on the iron overload severity.
The muscles and kidneys may appear hyperintense, due to iron deposition.
The spleen is characteristically spared and unaffected in hemochromatosis. In contrast, the spleen is almost always affected in hemosiderosis.
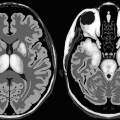
In superficial brain siderosis, a characteristic band of low T1 and T2 signal intensity is seen around the brain tissue, especially the pons, due to its location within the pontine cistern (Fig. 9.1.2).

Fig. 9.1.2
Axial T1W (a) and T2W (b) brain MRI show a hypointense rim that surrounds the pons (arrowhead) due to superficial brain siderosis
Further Reading
Argyropoulou MI, et al. MRI evaluation of tissue iron burden in patients with β-thalassemia major. Pediatr Radiol. 2007a;37:1191–200.
Bonetti MG, et al. Hepatic iron overload in thalassemic patients: proposal and validation of an MRI method of assessment. Pediatr Radiol. 1996;26:650–6.
Brasch RC, et al. Magnetic resonance imaging of transfusional hemosiderosis complicating thalassemia major. Radiology. 1984;150:767–71.
Chen CH, et al. Idiopathic pulmonary hemosiderosis: favorable response to corticosteroid. J Chin Med Assoc. 2008;71:421–4.
Deugnier Y, et al. Iron and the liver: update 2008. J Hepatol. 2008;48:S113–23.
Flyer MA, et al. Transfusional hemosiderosis in sickle cell anemia: another cause of an echogenic pancreas. Pediatr Radiol. 1993;23:140–2.
Koçak R, et al. The liver siderosis in beta-thalassemia intermedia and hemoglobin disease. J Islamic Acad Sci. 1993;6:42–5.
Positano V, et al. Improved T2* assessment in liver iron overload by magnetic resonance imaging. Magn Reson Imaging. 2008a;27:188–97. doi:10.1016/j.mri.2008.06.004.
Rosenberg W. Haemochromatosis Med. 2007;35:89–92.
Rosenberg W, et al. Haemochromatosis. Medicine. 2002;30:63–4.
9.2 β-Thalassemia Major (Cooley’s Anemia)
β-thalassemia major is a hereditary hemolytic anemia, characterized by deficiency in the hemoglobin beta chain synthesis. Patients with β-thalassemia major are prone to repeated attacks of intravascular hemolysis that requires repeated hospitalization and blood transfusions.
Patients with β-thalassemia major present with microcytic hypochromic anemia with signs of fatigue and cardiac tachycardia.
Repeated blood transfusion predisposes to hemosiderosis and tissue iron burden, which is the most severe complication of this disease. Hemosiderosis causes cardiomyopathy, hepatic failure, hypogonadism (pituitary siderosis), and endocrinal abnormalities. Bronze diabetes is a term used to describe diabetes mellitus induced in a patient with thalassemia due to pancreatic hemosiderosis. The term “bronze” refers to skin darkening and hyperpigmentation that is seen in patients with chronic thalassemia, due to deposition of hemosiderin in the subcutaneous tissues. Hypoparathyroidism is one of the most important endocrinal complications of thalassemia.
Extramedullary hematopoiesis is commonly observed in these patients due to increased body demands. Extramedullary hematopoiesis can be appreciated on plain radiographs as abnormally widened, flat bones.
Within the past few years, MRI has emerged as a powerful diagnostic tool to detect hemosiderosis through the body. Techniques for tissue iron burden quantification are well established for the liver and the heart. Early treatment with chelating agents (e.g., desferrioxamine) reduces the severity of the iron burden complication. MRI iron burden quantification helps in monitoring chelation therapy.
Signs on Skeletal Radiograph
Skull hair–on–end appearance: increase of the trabeculae within the skull bones, due to extramedullary hematopoiesis that widens the calvarial flat bones (Fig. 9.2.3)
Square-shaped metacarpals and thin cortex, due to bone marrow proliferation (Fig. 9.2.4)
Dilated ribs due to extramedullary hematopoiesis (Fig. 9.2.5)
Splaying of the femoral metaphysis (Erlenmeyer flask deformity)

Fig. 9.2.3
A plain radiograph of the lateral skull of a patient with thalassemia shows hair-on-end-appearance

Fig. 9.2.4
A plain radiograph of the hand shows squaring and expansion of the phalanges due to extramedullary hematopoiesis in a young patient with thalassemia

Fig. 9.2.5
AP plain radiograph shows expansion of the ribs due to extramedullary hematopoiesis (arrowheads)
Signs on CT
Hepatic hemosiderosis is one of the main causes of high-density liver on nonenhanced CT images (Fig. 9.2.6). The liver density will show high HU difference compared to the muscles and the spleen, with a range of 80–140 HU. Other causes of nonenhanced CT high-density liver are Wilson’s disease due to copper deposition, and hepatic iodine deposition, rarely seen in amiodarone toxicity.
Hepatosplenomegaly often occurs due to extramedullary hematopoiesis (Fig. 9.2.7).
Thoracic paraspinal masses and enlarged lymph nodes may be seen due to extramedullary hematopoiesis (Fig. 9.2.8).
Cerebral calcification may be seen due to hypoparathyroidism in thalassemia patients. Patients may uncommonly show bilateral symmetrical basal ganglia calcifications (Fig. 9.2.9).

Fig. 9.2.6
Axial nonenhanced CT of the abdomen shows high-density liver

Fig. 9.2.7
Axial nonenhanced CT of the abdomen shows hepatomegaly with left liver lobe hypertrophy

Fig. 9.2.8
Axial nonenhanced CT illustration of the thorax shows bilateral paraspinal masses due to extramedullary hematopoiesis (arrowheads). These masses can be easily mistaken for tumors. Other signs of extramedullary hematopoiesis support the diagnosis

Fig. 9.2.9
Axial nonenhanced brain CT shows bilateral, almost symmetrical, basal ganglia calcifications
Signs on MRI
Iron burden quantification is done with the use of T2* sequences (Fig. 9.2.10). The superparamagnetic properties of the iron deposited within the tissues cause decreased signal intensity of the tissues containing iron. As a result, hepatic parenchyma, splenic parenchyma, and cardiac muscles with siderosis appear hypointense compared to normal parenchyma.
In mild liver siderosis, the liver appears hypointense only on T2* images, compared to muscles. In moderate to severe cases, the liver appears hypointense to spleen and muscles in all sequences. The spleen shows almost the same picture as the liver, due to iron deposition. In primary hemochromatosis, only the liver shows decreased signal intensity, while the spleen is spared.
In the heart, iron overload appears as a dark ring on T1W, T2W, and T2* images.
In bronze diabetes, the pancreas and sometimes the adrenals show low signal intensity due to iron deposition.
In hypogonadism and signs of pituitary failure, the anterior pituitary shows low signal intensity on T1W, T2W, and T2* images, reflecting severe iron deposition.

Fig. 9.2.10
Axial abdomen section in different sequences illustrates the method of liver iron burden quantification in the liver and the paraspinal muscles
Further Reading
Argyropoulou MI, et al. MRI evaluation of tissue iron burden in patients with β-thalassemia major. Pediatr Radiol. 2007b;37:1191–200.
Drakonaski E, et al. Adrenal glands in beta-thalassemia major: magnetic resonance (MR) imaging features and correlation with iron store. Eur Radiol. 2005;15:2462–8.
Karimi M, et al. Prevalence of hepatosplenomegaly in beta thalassemia minor subjects in Iran. Eur J Radiol. 2007;59:120–2. doi:10.1016/j.ejrad.2007.09.027.
Karimi M, et al. Hypoparathyroidism and intracerebral calcification in patients with beta-thalassemia major. Eur J Radiol. 2008;70:481–4. doi:10.1016/j.ejrad.2008.02.003.
Lal A, et al. Focal splenic lesions as a cause of extramedullary hematopoiesis in a case of thalassemia. Eur J Radiol Extra. 2008;68:e125–7.
Louis CK. Low growth of children with β-thalassemia major. Indian J Pediatr. 2005;72:159–64.
Mavrogeni S, et al. Magnetic resonance evaluation of liver and myocardium iron deposition in thalassemia intermedia and β-thalassemia major. Int J Cardiovasc Imaging. 2008;24:849–54.
Papakonstantinou O, et al. MR imaging of spleen in beta-thalassemia major. Abdom Imaging. 2006;40:2777–82. doi:10.1007/s00261.006.9138-4.
Positano V, et al. Improved T2* assessment in liver iron overload by magnetic resonance imaging. Magn Reson Imaging. 2008b;27:188–97. doi:10.1016/j.mri.2008.06.004.
9.3 Sickle Cell Disease
Sickle cell disease (SCD) is an autosomal recessive genetic disorder, characterized by episodic attacks of hemolytic anemia and vaso-occlusive attacks due to “sickling” of the red blood cells (RBCs) under certain body conditions that include dehydration, metabolic acidosis, and low oxygen saturation.
SCD results from abnormal production of hemoglobin (Hb-S). Sickle cell patients are homozygous (HbSS), while heterozygous patients have “sickle cell trait.” SCD can also arise when Hb-S is combined with abnormal hemoglobin (e.g., Hb-S-thalassemia). Hb-S differs from the normal Hb-A only in the substitution of valine for glutamic acid in the sixth position of the β chain.
In SCD, the normal, discoid RBCs shape is transformed into a sickle-shaped, sticky mass during deoxygenation. These sickle cells can stick together, forming a hard mass that may lead to embolization and arterial infarction in different parts of the body.
Approximately 50 % of patients with SCD experience painful crises by the age of 5 years. Patients with sickle cell disease have natural protection against malaria; the reasons are unknown.
The Lungs in SCD
Patients with SCD have greater susceptibility to pneumonia (100 times more than other children), due to impaired immune status. The infective agents are commonly Streptococcus pneumoniae, Haemophilus influenzae, and Salmonella. Acute chest syndrome (ACS) is a term used to describe newly developed pulmonary consolidation, accompanied by fever, chest pain, dyspnea, and cough. The underlying cause is known and presumably due to fat emboli. ACS is the second most common cause for hospital admissions in children with SCD, after painful crises. ACS is also seen in up to 10 % of SCD patients after general anesthesia.
The Skeletal System in SCD
Skeletal manifestations in SCD range between vaso-occlusive crises, extramedullary hematopoiesis, osteomyelitis, and vertebral changes. Bone infarction is the most common cause of pain crises in SCD. However, silent infarctions do exist in patients with SCD. There are four zones seen in bone infarction by histology: the zone of cell death located in the center, the zone of ischemic tissue, the zone of hyperemia, and the outer zone of normal bony tissue.
Osteomyelitis means inflammation of the bone and the bone marrow. Osteomyelitis is commonly caused by Salmonella infection in sickle cell patients. In nonsickle cell patients, the most common cause of osteomyelitis is Staphylococcus aureus.
Differential Diagnoses and Related Diseases
Hand–foot syndrome is an uncommon disease seen in sickle cell patients in up to 20 % of cases, characterized by bilateral inflammation and swelling of the fingers and toes (dactylitis). Patients present with fever, bilateral digital swelling in the hands and feet, leukocytosis, and pain. It can be mistaken for osteomyelitis in the initial presentation. Osteomyelitis is uncommonly known to cause bilateral infection in the hands and feet simultaneously. Also, osteomyelitis often involves the long bones, not the small bones of the hands and feet. Most patients experience this syndrome before 4 years of age, and the disease has not been reported beyond 7 years. The condition is self-limiting, with a duration that varies from a week to a month.
What Is the Difference Between Osteonecrosis, Avascular Necrosis, and Bone Infarction?
Osteonecrosis is ischemic death of the bone and bone marrow.
Avascular necrosis is osteonecrosis that occurs in the epiphyses.
Bone infarction is osteonecrosis that occurs in the metaphyses or diaphyses.
The Brain in SCD
In SCD, the brain may be damaged due to infarction from sickle cell emboli or from vasculitis. Up to 25 % of patients with SCD will have a neurological complication over their lifetime; 11 % of these complications will occur by the age of 20 years. Silent infarction is defined as MRI manifestation of cerebral infarction in the absence of clinical symptoms and occurs in up to 22 % of patients with SCD.
The Spleen in SCD
Multiple spleen infarctions due to vaso-occlusive crises are a very common feature in SCD. With time, the spleen is replaced by fibrous tissue and by calcium and hemosiderin deposition (called autosplenectomy). Up to 94 % of patients are asplenic by the age of 5 years.
Patients with splenectomy are susceptible to infection with Staphylococcus pneumoniae, Salmonella, and Haemophilus influenzae. Pneumococcal vaccine is often started between 2 and 5 years of age.
Sequestration syndrome is another condition that commonly occurs in SCD patients, characterized by rapid pooling of the blood within the spleen, resulting in intravascular volume depletion and dropping hematocrit levels. When the sequestration is severe, patients present with abdominal fullness, thirst, tachycardia, and tachypnea that may rapidly progress into circulatory collapse. Up to 30 % of patients experience sequestration syndrome between the ages of 6 months and 3 years.
Signs on Chest Radiograph
Pneumonia is seen as areas of patchy lung infiltration with air bronchogram. The airspace disease may be lobar or diffuse.
Acute chest syndrome is seen as single or multiple patchy areas of airspace disease, often confined to the middle and lower lobes. Up to 60 % of patients with ACS show normal chest radiograph.
Signs on Skeletal Radiograph
Bone infarction: it is seen as a radiolucent area surrounded by the sclerotic rim, typically in the epiphyses and the medullary cavity (Fig. 9.3.11). Later, sclerosis of the infarcted areas causes the appearance of dense bone within the affected bone (bone-in-bone appearance).
H–shaped vertebra: there is central end plate depression, with sparing of the anterior and posterior margins due to previous infarctions of vertebral bodies. An H-shaped vertebra is a characteristic sign of SCD (Fig. 9.3.12).
Expansion of the diploic medullary spaces of the skull due to increased hematopoietic demands (hair-on-end appearance).
Osteomyelitis: the early changes seen radiographically are soft-tissue swelling or a mass, occasionally gas, periosteal reaction, and (later) cortical destruction. There are usually no signs on plain radiograph in the first 2 weeks of infection. The cortical destruction first appears as small lucent holes (permeative destruction), followed later by larger coalescent lesions (moth-eating destruction). Osteomyelitis can be difficult to differentiate from infarction.
Hand–foot syndrome: the typical signs of dactylitis include soft-tissue swelling of the digits, cortical thinning, multiple intramedullary radiolucent deposits, and thick periosteal new bone formation (Fig. 9.3.13). The radiological manifestations are completely reversible after 8 months.
Protrusio acetabuli: it may occur in SCD in up to 20 % of cases.

Fig. 9.3.11
Plain radiograph of the distal femur metaphysis shows radiolucent areas surrounded by sclerotic rims (arrowheads) due to old bone infarction in a patient with sickle cell disease (SCD)

Fig. 9.3.12
Lateral vertebral plain radiograph of a patient with SCD shows H-shaped thoracic vertebra

Fig. 9.3.13
Plain radiograph of both hands in a sickle-cell patient shows thick periosteal new bone formation affecting the fifth and fourth right metacarpal bones and the fifth left metacarpal bone (arrowheads), changes indicating hand-foot syndrome
Signs on Chest CT
Extramedullary hematopoiesis may be found as bilateral or unilateral, smooth or lobulated paraspinal masses in the lower thoracic spine, without vertebral erosions. History of hematological disease is the key diagnosis to differentiate these masses from tumors.
Signs on Abdominal CT
Spleen infarction is seen on noncontrast-enhanced images as a hypodense, wedge-shaped area, which typically starts from the periphery toward the center, with no contrast enhancement (Fig. 9.3.14).
Sequestration syndrome is seen as splenomegaly with hypodense peripheral areas.

Fig. 9.3.14
Axial abdominal postcontrast CT shows multiple hypodense wedge-shaped areas within the spleen, due to multiple areas of infarction
Signs on Skeletal MRI
MRI is important in the early detection of bone infarction. On T2W images, there is an area surrounded by a hyperintense line and an outer hypointense line (double–line sign). The high-intensity line represents the zone of hyperemia. Double-line sign is found in up to 80 % of cases of bone infarction.
The yellow marrow is made of 80 % fat, 15 % water, and 5 % proteins. On MRI, it gives high signal in T1W images. In contrast, the red marrow is made of 40 % fat, 40 % water, and 20 % proteins. This high water content gives low signal in both T1W and T2W images. Extracellular hematopoiesis, especially within the vertebrae, can be suggested by observation of the intervertebral disk signal. Normally, the vertebral bodies have higher signals than the intervertebral disks on T1W images, due to the fatty marrow. In extracellular hematopoiesis, the yellow marrow is reconverted into red marrow due to the hematopoietic demands, resulting in low signal intensity of the vertebral bodies compared to the intervertebral disks on T1W images (high–density disk sign). This sign is observed in any disease with bone marrow infiltration.
Signs on Brain MRI
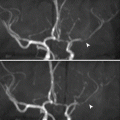
Silent infarction is detected as a high signal intensity lesion within the white matter on T2W or FLAIR images.
Moyamoya disease is detected by its classical “puff of smoke” appearance on MR angiography and occlusion of the ipsilateral internal carotid artery.
Further Reading
Babhulkar SS, et al. The hand-foot syndrome in sickle-cell haemoglobinopathy. J Bone Joint Surg (Br). 1995;77-B:310–2.
Ejindu VG, et al. Musculoskeletal manifestations of sickle cell disease. Radiographics. 2007;27:1005–21.
Janet Watson R, et al. The hand-foot syndrome in sickle-cell disease in young children. Pediatrics. 1963;31:975–82.
Lonergan GJ, et al. Sickle cell anemia. Radiographics. 2001;21:971–94.
Lukens JN. Sickle cell disease. Dis Mon. 1981;27:1–56.
Lukens JN. Sickle cell disease. Dis Mon. 1981;27:1–56.
Schatz J, et al. Sickle cell disease as a neurodevelopmental disorder. Ment Retard Dev Disabil Res Rev. 2006;12:200–7.
9.4 Pernicious Anemia
Pernicious anemia (PA) is a disease characterized by the development of megaloblastic anemia due to destruction of the gastric parietal cells. Megaloblastic anemias are a subgroup of macrocystic anemias (large volume red blood cells), which most commonly arise due to vitamin B12 and folate deficiencies.
Normally, the parietal cells in the gastric mucosa secrete an intrinsic factor, which is important for absorption of vitamin B12 from the gastrointestinal tract. The fundus of the stomach contains parietal cells, the body contains the cells responsible for the secretion of pepsin and hydrochloric acid, and the antrum contains G cells. Patients with PA develop antigastric parietal cells (GPC) and anti-intrinsic factor antibodies (gastric parietal cells antibodies, or AGPA), which attack the parietal cells and cause autoimmune gastritis. Destruction of the gastric parietal cells leads to gastric mucosal atrophy and compromises the production of the intrinsic factor. Moreover, the atrophic gastritis also compromises the gastric acid pump, leading to deficiency in the secretion of gastric acids (hypo- or achlorhydria). Loss of vitamin B12 causes defective synthesis of the bone marrow cellular activities, leading to the development of megaloblastic erythropoiesis and macrocytic anemia.
Chronic gastritis induces enterochromaffin-like cell hyperplasia, which may result in the development of gastric carcinoid tumors. Compared to the general population, gastric adenocarcinoma is 3–5 times more frequent among patients with PA. The activity of natural killer cells, which participate in immunosurveillance against tumor dissemination, is believed to be compromised in patients with PA.
Atrophic gastritis is divided into two types. Atrophic gastritis type (a) is characterized by mucosal atrophy affecting the fundus and the body of the stomach, with antral sparing. In contrast, atrophic gastritis type (b) is characterized by antral mucosal atrophy, with limited involvement of the fundus and body.
AGPAs are found in 20 % of patients with diabetes mellitus type 1 (DMT1). Researchers suggest that patients with DMT1 should be regularly screened for atrophic gastritis. PA is rare found in the general population, with an incidence of 0.1–2 % of the population. On the other hand, patients with DMT1 have a threefold increased incidence of PA, with a prevalence of 2.5–4 %.
Diagnosis is confirmed by detecting AGPA levels in the serum by the Schilling test.
Signs on Barium Meal
In a normal double-contrast barium meal examination, the stomach mucosal folds (rugae) are observed arranged in an irregular fashion through the fundus, body, and antrum (Fig. 9.4.15). At the lesser curvature near the pylorus, the mucosal folds become longitudinal and are known as magenstrasse. Area gastrica is an area of nodular mucosal elevation located near the antrum (Fig. 9.4.16).

Fig. 9.4.15
Double-contrast barium meal shows the normal configuration of the stomach with the mucosal folds (rugae) nicely demonstrated

Fig. 9.4.16
Double-contrast barium meal shows area gastrica, seen as an area with nodular mucosal pattern
In Pernicious anemia, there is a tubular-shaped fundus <8 cm in diameter absent or reduced amount of the normal gastric rugae in the fundus of the body (bald fundus) and small or absent area gastrica (Fig. 9.4.17).

Fig. 9.4.17
Double-contrast barium meal of a patient with pernicious anemia demonstrates severe atrophic gastritis with loss of the normal mucosal folds. Compare this image with Fig. 9.4.15
Further Reading
Levine MS, et al. Atrophic gastritis in pernicious anemia: diagnosis by double-contrast radiography. Gastrointest Radiol. 1989;14:215–9.
Tzellos TG, et al. Pernicious anemia in a patient with type 1 diabetes mellitus and alopecia areata universalis. J Diabetes Complications. 2009;23:434–7. doi:10.1016/j.jdiacomp.2008.o5.003.
Vargas JA, et al. Natural killer cell activity in patients with pernicious anemia. Dig Dis Sci. 1995;40:1538–41.
Varis K, et al. An appraisal of tests for severe atrophic gastritis in relatives of patients with pernicious anemia. Dig Dis Sci. 1979;24:187–91.
Wickramasinghe SN. Diagnosis of megaloblastic anemias. Blood Rev. 2006;20:299–318.
9.5 Hemophilia
Hemophilia is a rare, chronic X-linked genetic disease, characterized by the body’s inability to form clotting factors necessary for the blood clotting cascade to occur, resulting in a tendency toward spontaneous bleeding or bleeding after minor body trauma.
The word hemo means bleeding, and the word philia means tendency toward something. Patients with hemophilia have a tendency for slow bleeding, at a constant rate and without clotting, into muscles, joint spaces, and body cavities.
Blood clotting is a complicated process that involves three primary steps. The first step involves immediate constriction of the blood vessels in the area of injury. The second step involves the formation of a platelet plug that stops the bleeding. The third step involves the activation of 12 clotting factors (identified by Roman numerals) that transform the platelet plug into a more stable clot by transforming it into fibrin. The activation of clotting factors is referred to as the “clotting cascade,” because each factor stimulates the next factor in the series, until the formation of the fibrin. Deficiency of one factor will stop the cascade, and a stable clot will not form.
There are three types of hemophilia:
Hemophilia A results from deficiency of clotting factor VIII, and it is the most common form of hemophilia (80 %). The incidence is 1:10,000 people.
Hemophilia B (Christmas disease) results from deficiency of clotting factor IX and constitutes up to 23 % of hemophilia cases. The disease was named after a young boy, Stephen Christmas, who was the first patient identified with this disease. The incidence is 1:40,000 people.
Hemophilia C results from deficiency of clotting factor XI. It is a much rarer form and constitutes less than 2 % of all cases of hemophilia.
Patients with hemophilia are prone to slow, steady, and continuous bleeding after minor trauma. Bleeding can also occur spontaneously without trauma. The most important complications include bleeding into joints (hemarthrosis), internal bleeding, intracranial bleeding, and susceptibility from hematological infections due to recurrent blood transfusions.
Bleeding into the joints can occur in any joint, but it commonly affects the knees and the elbows. Target joint is a term used in hemophiliacs to indicate a joint with more frequent bleeding than other joints, commonly the knee. The joint synovium is rich in blood vessels, causing it to bleed easily. Multiple bleeding within the joint causes synovium hypertrophy, which later causes articular joint destruction and osteoarthritis. Patients with joint bleeding experience severe pain, due to swelling of the affected joint with stretching of the intra-articular structures by the entrapped blood. Recurrent joint bleeding can stimulate the growth plate, resulting in bony hypertrophy.
Bleeding into the muscles (e.g., the psoas muscle), if not controlled, may lead to muscular swelling, nerve damage, and development of compartment syndrome. Hemophilic pseudotumor is a rare complication of hemophilia, occurring in 1–2 % of hemophiliacs. It results from a chronic, encapsulated, slow-growing intramuscular hematoma that displaces the surrounding tissues. Limb enlargement, bone resorption, and muscle and skin necrosis all can be seen in severe cases.
Internal bleeding can be seen as skin bruising, nose bleeding, or blood in the urine (hematuria). Moderate hemophiliacs may bleed 5–6 times per year. Severe hemophiliacs may have 2–3 bleeding episodes per month.
Intracranial bleeding may occur within the brain parenchyma or within the subarachnoid space. Altered consciousness, headache, nausea, and vomiting in a patient with hemophilia after a minor head injury should be considered intracranial bleeding and investigated with a head CT without delay.
Myositis ossificans (MO), also known as “Sterner’s tumor,” is a rare, nonneoplastic condition characterized by formation of bone within muscles. The disease may be hereditary (fibrodysplasia ossificans progressiva, Munchmeyer disease), nontraumatic (e.g., in hemophilia), or traumatic, which is the most common form (e.g., after muscle trauma). The previous classification is applied to intramuscular MO; however, MO can arise against a bone (parosteal MO) or evolve as periostitis (periosteoma).
In the early stages of MO, there are richly vascularized fibroblastic cell proliferations with prominent mitotic activity that mimic malignancy (early pseudosarcomatous phase). As the cells mature, the lesion typically shows three distinct zones. The first zone is composed of rapidly proliferating fibroblasts with areas of hemorrhage and necrosis; the intermediate layer is composed of osteoblasts with osteoid matrix with islands of endochondral ossification; the third outer zone is composed of mature bone, separated from the surrounded tissue by myxoid-fibrous tissue. The peripheral zone usually calcifies at 6–8 weeks after lesion initiation, and complete lesion ossification can be seen 5–6 months from the onset of symptoms. Up to 30 % of lesions regress and resolve spontaneously with maturation.
Patients with MO typically present with painful swelling, commonly in the lower limbs (60–75 % of cases). Patients, especially children, may not recall the incidence of trauma. Diagnostic imaging approach for a patient with painful swelling, with suspicion of MO, should start with conventional radiography, US, CT, and later MRI, as the MRI appearance of MO is generally nonspecific unless the lesion starts to mature. History of trauma is important to suspect MO; however, the absence of history of trauma does not exclude it.
Differential Diagnoses and Related Diseases
Von Willebrand’s disease is a bleeding disorder that mimics hemophilia and results from deficiency of von Willebrand factor.
Signs on Radiographs
Enlargement of the epiphysis (100 %), joint swelling with soft-tissue swelling (81 %), and osteoporosis (95) are commonly found in hemophilic arthropathy. Reduction of the joint space and signs of osteoarthritis are also commonly found (Fig. 9.5.18).
Genu recurvatum is a disabling deformity condition, characterized by hyperextension of the knee to >5°. This deformity may occur in patients with hemophilia after recurrent knee hemarthrosis.
Hemophilic pseudotumor is seen as an expanding limb with soft-tissue mass and lytic destruction of the bone within the mass. Bones that are often affected by pseudotumors are the femur, tibia, pelvis, and bones of the hands.
MO is detected classically as bone within areas of soft tissue. The calcification is typically peripheral with a radiolucent center depending on the level of maturation. This pattern of ossification is important to differentiate MO from osteosarcoma, which typically shows a dense center and sunray peripheral edges. The ossification may appear as nonspecific flocculent areas of soft-tissue calcification called “dotted veil pattern” or may characteristically follow the course of muscle fibers (Fig. 9.5.19).

Fig. 9.5.18
Anteroposterior (a) and lateral (b) plain knee radiographs in a patient with hemophilic arthropathy. Notice the knee with obvious osteoarthritis, sclerosis, and joint effusion (arrowheads)

Fig. 9.5.19
Anteroposterior bilateral radiograph of the legs and distal femur shows bilateral calcification that involved the vastus medialis and the rectus femoris muscles in a patient with myositis ossificans. Notice how the calcification follows the muscle fibers
Signs on US
In the early stage of myositis ossificans, the mass is detected as a hypoechoic mass with an outer hypoechoic zone enclosing a broader hyperechoic zone, which again encloses a central hypoechoic zone. After maturation, the outer layer becomes hyperechoic due to ossification.
Signs on MRI
In general, the MRI findings of MO are nonspecific; however, a peripheral rim with low T1 and T2 signal intensities surrounding a heterogeneous intramuscular mass can be a clue for MO. The dark rim represents the calcified peripheral zone. It should be remembered that, at the initial stage, MO resembles musculoskeletal sarcomas, even when a biopsy is done.
After contrast injection, MO shows peripheral rim enhancement in the early stages, which can lead to mistaking it for an abscess or a necrotic tumor.
Further Reading
Bae DK, et al. Total knee arthroplasty in hemophilic arthropathy of the knee. J Arthroplasty. 2005;20:664–8. doi:10.1016/j.arth.2005.01.008.
Dauty M. Iliopsoas hematoma in patients with hemophilia: a single-center study. Joint Bone Spine. 2007;74:179–83.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree