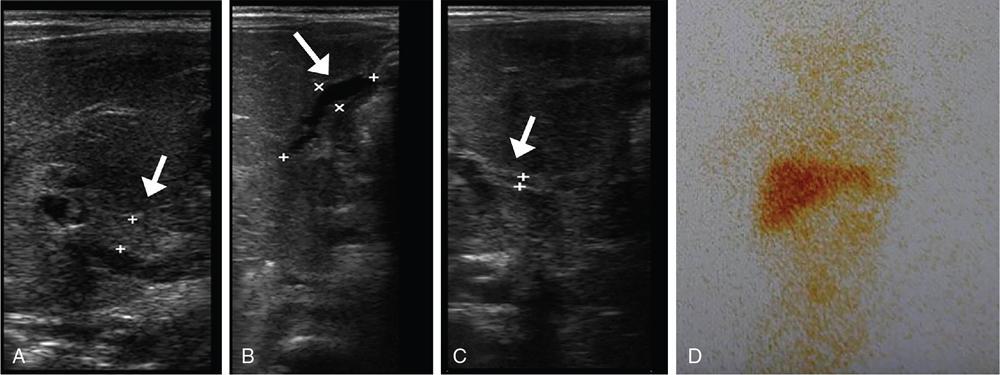
Subramaniyan Ramanathan, Vineetha Raghu, Tahiya Salem Alyafei, Mahmoud Al Heidous Congenital hepatobiliary anomalies include a variety of conditions often presenting with neonatal jaundice early in life or portal hypertension later in life, and some of them can be asymptomatic and incidentally discovered in adults. They may be due to several hereditary or developmental disorders. Physiological jaundice in the neonatal period typically occurs at 3–7 days, and bilirubin levels are <12 mg/dL in full term and <14 mg/dL in preterm neonates. The onset of jaundice before 24 hours, the presence of jaundice persisting beyond 2 weeks, exceeding the normal range, or the presence of conjugated hyperbilirubinemia are abnormal. In this chapter, we discuss in detail the various imaging features with a brief note on the clinical presentation, pathogenesis and management of biliary atresia, Alagille syndrome, Caroli’s disease, choledochal cysts, congenital hepatic fibrosis, polycystic liver disease and biliary hamartoma. Biliary atresia results from severe to complete obliteration of the extrahepatic biliary tree, and accounts for one-third of the cases of persistent neonatal jaundice. Its incidence varies from 1:10,000 to 1:25,000 births with higher rates among Asian population. The possible aetiopathogenesis of this condition is thought to be perinatal foetal cholangitis with or without a malformed biliary tree. The two most common causes of neonatal cholestasis include biliary atresia and neonatal hepatitis, and they together encompass 60%–90% of cases. Although the clinical and biochemical features are very similar in these two conditions, their management is different and imaging plays a central role in differentiating these closely related conditions. Prompt diagnosis and urgent surgical management may help prevent cirrhosis and permanent liver damage. This is owing to the fact that small patent biliary ductules are present at the hepatic hilum at birth which gradually fibrose and disappear by 3–4 months. This condition is associated with situs inversus; congenital heart disease; trisomies 17, 18, 21 and choledochal cysts. Polysplenia is the most common extrahepatic anomaly associated with biliary atresia described as biliary atresia splenic malformation (BASM) and found in 20% of the patients. Clinically, the child presents with signs of obstructive jaundice, including yellowish discolouration of skin and mucosa, dark urine and pale stools. Hepatomegaly may be present. Cirrhosis and portal hypertension may develop if untreated. Laboratory investigations reveal conjugated hyperbilirubinemia, elevated alkaline phosphatase, gamma-glutamyl transpeptidase and serum aminotransferases. Biliary atresia can be categorized into three types based on the extent of biliary involvement, known as the Kasai classification (Table 9.7.1) US is the first screening test and is usually requested to distinguish biliary atresia from neonatal hepatitis. Various findings have been described and can be categorized into primary and secondary features. Primary features include triangular cord sign, abnormal gallbladder, poor postprandial contractility and absent common bile duct (CBD). Hepatomegaly, splenomegaly and enlarged right hepatic artery are the secondary findings. The triangular cord sign has been described as a triangle-shaped or tubular echogenic structure measuring more than 3 mm seen anterior to the portal vein; this sign, although highly specific, has a low sensitivity. The gallbladder ‘ghost triad’ has been described in biliary atresia as gallbladder with length less than 1.9 cm, indistinct wall with lack of complete echogenic mucosal lining and irregularity of its outline. The Lee criterion refers to the thickness of anterior wall of the right portal vein measuring more than 4 mm. Other findings include enlarged hepatic artery >1.5 mm and ratio of hepatic artery to portal vein diameter >0.45. Overall, triangular cord sign shows the highest diagnostic accuracy with a reported sensitivity of 74% and specificity of 97%. The combination of gallbladder abnormalities and triangular cord sign can be used to increase the sensitivity to 95% (Fig. 9.7.1). Hepatobiliary scintigraphy (Hepatobiliary iminodiacetic acid or HIDA, 99mtechnetium mebrofenin, diisopropyl-iminodiacetic acid or the DISIDA scan), endoscopic retrograde cholangiopancreatography (ERCP) and intraoperative cholangiography are further investigations which aid in the diagnosis. The HIDA scan reveals liver uptake with biliary nonexcretion of the radioisotope into the bowel at 24 hours, and has a sensitivity approaching 100%. However, it has a lesser specificity of 75%–80%. Severe hepatocellular dysfunction may also lead to nonexcretion of tracer into the gut and may confound the diagnosis. Magnetic resonance cholangiopancreatography (MRCP) can be used to demonstrate the intrahepatic ducts and CBD in equivocal cases, thereby ruling out biliary atresia. However, it is a technically difficult examination to perform in neonates due to the need for sedation, cost and availability issues. Although both false positives and false negatives have been reported, limited available studies show good diagnostic accuracy with one recent study reporting approximate sensitivity of 97% and specificity of 95% for the MRI triangular cord sign. If the diagnosis could not be reached on imaging, liver biopsy can be useful in a certain subset of patients. Last resort for definitive diagnosis is intraoperative cholangiogram and it remains the gold standard. Portoenterostomy (Kasai procedure) is the surgical treatment of choice; however, it is unlikely to be of value if performed after the child is 3 months old. Later in the course of the disease, hepatic transplantation is the treatment of choice. Alagille syndrome is a rare hereditary condition characterized by chronic cholestasis and has an autosomal dominant transmission with variable penetration. However, about 50% of the cases are sporadic, occurring due to de novo mutations involving the JAG1 or NOTCH2 gene. It is an important cause of familial neonatal cholestasis with an incidence of 1 per 100,000 live births. In contrast to biliary atresia which predominantly involves the extrahepatic biliary tree, there is hypoplasia/paucity of the interlobular biliary ducts (PIBD). It may also present with abnormal facies, ocular abnormalities, hepatosplenomegaly, vertebral anomalies, peripheral pulmonary stenosis and cardiac malformations. Approximately 15% of these patients go on to develop cirrhosis and liver failure. Intracranial haemorrhage and moyamoya-like condition resulting from associated vascular anomalies may account for a significant percentage of mortality and morbidity. Differential diagnoses include biliary atresia, neonatal hepatitis and progressive familial intrahepatic cholestasis (Byler’s disease). Other disorders causing bile duct paucity include alpha-1-antitrypsin deficiency, cystic fibrosis and Zellweger syndrome amongst others. Imaging features depend on disease severity and include hepatomegaly, periportal fibrosis/cirrhosis, and splenomegaly, or sometimes, a normal liver. This condition needs to be differentiated from biliary atresia, as Kasai procedure is not useful and can also be detrimental in Alagille syndrome. Small gallbladder on US and nonvisualization of CBD on MRCP are reported in both BA and Alagille and cannot be used to differentiate them. However, triangular cord sign and hepatic artery enlargement are absent in Alagille syndrome and features of portal hypertension are less frequent. Nuclear scintigraphy (HIDA scan) findings are similar to biliary atresia with no bowel excretion of the isotope. Liver biopsy and histopathology are confirmatory for diagnosis. The only available management is orthotopic liver transplantation, failing which these patients do not survive beyond the third decade. This is a spectrum of diseases of the liver and biliary system resulting from aberrations in the embryologic development of the ductal plates. Ductal plates are cylindrical layers of cells developing in the first to eighth week of gestation, surrounding a portal venous branch and eventually involuting partially (beginning at the twelfth week) to form the biliary ducts. Disturbances in this process of involution lead to ductal plate malformations. They encompass five important conditions, namely: The cysts in polycystic disease and biliary hamartoma do not communicate with the biliary radicles (as depicted on MRCP) indicating that they have lost communication with the biliary tree; whereas those in choledochal cysts, and Caroli’s disease do have a communication. The importance of these conditions is that, although they have varied clinical presentations, they all develop from ductal plate malformations in varying stages of development, and can coexist with each other as well as with other renal abnormalities. Cystic biliary atresia is a rare variant of biliary atresia characterized by cysts affecting the obliterated biliary tract. It is usually suspected in an infant presenting with cholestatic jaundice, with US demonstrating a cyst at the porta hepatis. It is an important differential of the more common choledochal cyst. It is important to differentiate these two conditions because they have differing management protocols. The presence of the triangular cord sign, associated gallbladder abnormalities, lack of intracystic calculi or sludge, absence of intrahepatic biliary dilatation and relatively smaller size of the cyst support the diagnosis of cystic biliary atresia over choledochal cyst. Choledochal cysts are characterized by varying degree and morphology of extra- and, sometimes, intrahepatic biliary dilatation. The incidence of these cysts follows a 4:1 female: male ratio with approximate incidence of 1–2:100,000–150,000 live births and more prevalent in Asian population. The possible aetiopathogenesis is an abnormal proximal insertion of the pancreatic duct into the CBD leading to a long common channel, reflux of pancreatic enzymes into the biliary tree, cholangitis, biliary obstruction and dilatation. The common channel measuring >1.5 cm long is described as the abnormal pancreaticobiliary junction (APBJ) and is reported in 96% of cases of choledochal cysts. Another theory is sphincter of Oddi dysfunction due to paucity of ganglion cells, leading to pancreatic reflux. Todani et al. have classified these cysts into five types based on their morphology (Table 9.7.2). A type VI cyst has also been described which refers to isolated dilatation of the cystic duct.
9.7: Hepatobiliary system: Congenital anomalies
Introduction
Biliary atresia
Type
Biliary Involvement
I
Only CBD
IIa
Only CHD
IIb
CHD with CBD and cystic duct
III
CHD, CBD, cystic duct and intrahepatic ducts up to porta hepatis
Alagille syndrome (alagille–watson syndrome)
Fibropolycystic liver disease
Choledochal cysts
Radiology Key
Fastest Radiology Insight Engine









