Description
Thrombosis
Non-atherosclerotic intimal lesions
Intimal thickening
Normal accumulation of smooth muscle cells (SMCs) in the intima in the absence of lipid or macrophage foam cells
Absent
Intimal xanthoma
Superficial accumulation of foam cells without a necrotic core or fibrous cap. Based on animal and human data, such lesions usually regress
Absent
Progressive atherosclerotic lesions
Pathologic intimal thickening
SMC-rich plaque with proteoglycan matrix and focal accumulation of extracellular lipid
Absent
Fibrous cap atheroma
Early necrosis: focal macrophage infiltration into areas of lipid pools with an overlying fibrous cap
Absent
Late necrosis: loss of matrix and extensive cellular debris with an overlying fibrous cap
Thin-cap fibroatheroma
A thin fibrous cap (<65 μm) infiltrated by macrophages and lymphocytes with rare or absence of SMCs and relatively large underlying necrotic core. Intraplaque hemorrhage/fibrin may be present
Absent
Lesions with acute thrombi
Plaque rupture
Fibroatheroma with cap disruption; the luminal thrombus communicates with the underlying necrotic core
Occlusive or non-occlusive
Plaque erosion
Plaque composition, as above; no communication of the thrombus with necrotic core. Can occur on a plaque substrate of pathologic intimal thickening or fibroatheroma
Usually non-occlusive
Calcified nodule
Eruptive (shedding) of calcified nodule with an underlying fibrocalcific plaque with minimal or absence of necrosis
Usually non-occlusive
Lesions with healed thrombi
Fibrotic (without calcification)
Fibrocalcific (±necrotic core)
Collagen-rich plaque with significant luminal stenosis. Lesions may contain large areas of calcification with few inflammatory cells and absence of necrosis. These lesions may represent healed erosions or ruptures
Absent
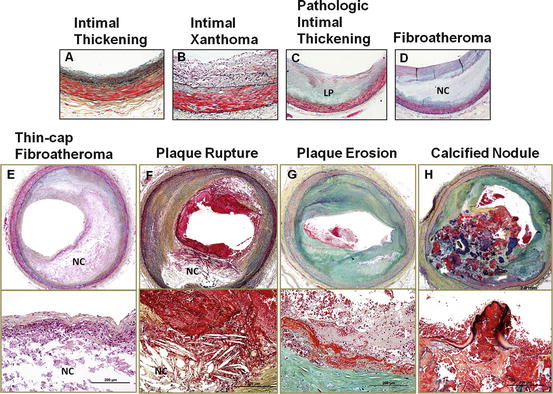
Fig. 2.1
Plaque atherosclerotic progression in human coronary arteries. These histologic images illustrate the various lesion morphologies of human coronary atherosclerosis. (a) Adaptive intimal thickening is present from birth and consists of a smooth muscle-rich intima. (b) Intimal xanthoma are predominantly foam cell-rich lesions that are found in the young, but known to regress in adults. (c) Pathologic intimal thickening (PIT) is the first of the progressive plaques marked by an acellular lipid pool rich in proteoglycan; inflammation in macrophages when present is typically confined to the most luminal aspect of this plaque. (d) Fibroatheroma (FA) are lesions with areas of necrosis characterized by cellular debris and cholesterol monohydrate with varying degrees of calcification or hemorrhage. (e) Thin-cap fibroatheroma (TCFA) or vulnerable plaques are recognized by their relatively large necrotic core and thin fibrous cap which is infiltrated by numerous macrophages. (f) Plaque rupture leads to exposure of necrotic contents to the blood flow, resulting in the triggering of the coagulation cascade. The luminal thrombus at the site of rupture is platelet rich (white thrombus). (g) Erosion is another entity that gives rise to coronary thrombosis. Erosions can occur on a substrate of PIT or FA. (h) Calcified nodules (a minor but viable mechanisms of thrombosis) depict eruptive fragments of calcium that protrude into the lumen causing a thrombotic event. LP lipid pool, NC necrotic core
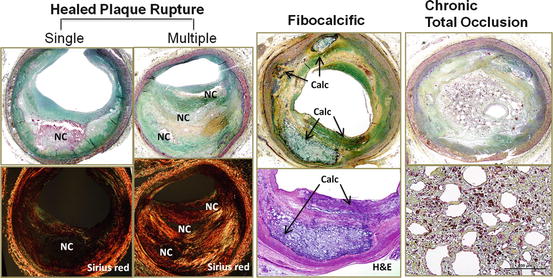
Fig. 2.2
Fate of atherosclerotic plaque. Boxes on the left show healed plaque ruptures where the newly formed fibrous cap is rich in proteoglycans and type III collagen (type III collagen appears green with Sirius red stain under polarized light) with interspersed smooth muscle cells. Repeated ruptures lead to a multilayered appearance with necrotic cores and overlying fibrous layers, resulting in severe luminal narrowing. Multiple healed plaque ruptures are thought responsible for progressive luminal narrowing. Fibrocalcific plaque (middle column) is thought to be a burnt-out lesion, which is associated with a calcified sheet of plaque matrix (arrows). Boxes on the right show histologic features of chronic total occlusion. The lumen is characterized by recanalized organized thrombus that is rich in proteoglycan matrix with presence of iron deposition and macrophages. Calc calcification, NC necrotic core
2.1 Adaptive Intimal Thickening and Intimal Xanthoma (Fatty Streaks)
The earliest manifestation of vascular change is “adaptive intimal thickening” (AHA Type I), which consists of several layers of smooth muscle cells (SMCs) in an extracellular matrix with no or little inflammatory cell infiltration. Intimal thickening is observed in 35 % of neonates, and the intima/media ratio at birth is 0.1 increasing progressively to reach 0.3 by 2 years of life [4]. This change is considered adaptive (non-atherosclerotic) since the SMCs exhibit a very low proliferative activity and exhibit an anti-apoptotic phenotype [5]. Although the adaptive intima increases in thickness with aging, very rarely does it grow into a disease compromising blood flow. The change of shear stress is a trigger for abnormal responses in the endothelial lining [6] as well as changes secondarily induced in SMC phenotype [7]; however, the detailed mechanism remain elusive.
“Fatty streak” or “Intimal xanthoma” (AHA Type II) is a lesion that is not raised and is primarily composed of abundant foamy macrophages interspersed between SMCs. Although the AHA classification alludes to this entity as the earliest lesion of atherosclerosis, from our experience and reports of human and animal studies, the lesion is reversible at least in some locations [8, 9]. Some reports suggest that the modification of extracellular matrix is responsible for the progression of macrophage infiltration implying a role for biglycan and decorin proteoglycans [10], but the mechanism remains largely uncertain.
2.2 Pathologic Intimal Thickening
“Pathologic intimal thickening” (PIT, AHA Type III) is recognized as the earliest progressive (irreversible) lesion by most research groups. The lesion is characterized by layers of proliferating SMCs near the lumen and an underlying lipid pool present at the intimal medial border. The origin of lipid pool is not fully understood. The area of the lipid pool is rich in proteoglycan versican and hyaluronan as well as extracellular lipid deposits but is devoid of SMCs and macrophages. It has been demonstrated that there is an affinity of the lipid pool to retain plasma lipoprotein, which suggests that the accumulation of extracellular lipid is likely derived from the influx of plasma lipoproteins [11]. Williams et al. proposed a “response-to-retention” hypothesis: the retention of atherogenic lipoprotein associated with the extracellular matrix such as proteoglycan versican and hyaluronan is an initiating event in early atherogenesis [12]. Recent studies reinforce this hypothesis by demonstrating that structural changes in the glycosaminoglycan chain of proteoglycans are an initial proatherogenic step that promotes the binding and retention of lipoproteins [13]. An alternative hypothesis suggests that the membranes of apoptotic SMC may be an alternative source for lipid in PIT [14]. Apoptotic SMCs within lipid pools are recognized by membrane remnants (cages of basal lamina) and the presence of microcalcification representing calcified mitochondria [15]. However, the proof supporting this mechanism remains speculative.
Another important hallmark of PIT is the presence of varying degrees of foamy macrophage accumulation near the luminal aspect of the plaque (apart from the lipid pool) albeit this does not necessarily apply to all cases. Lesions demonstrating PIT with foamy macrophages are considered a more advanced stage of atherosclerosis as reported by Nakashima et al. in their systematic study of early coronary plaques [16]. We believe that macrophages invade the plaque from the luminal surface. Although the precise nature of focal macrophage accumulation in PIT is not fully elucidated, it is speculated that retention or modified lipoprotein along with activation of vascular adhesion molecules like VCAM-1 and ICAM-1 expressed by endothelial cells stimulates the recruitment of macrophages [17, 18]. In addition, lesions with PIT exhibit varying degrees of free cholesterol represented by empty fine crystalline structures in paraffin-embedded sections that accumulate within lipid pools. Although it is assumed that free cholesterol originates from dead foam cells, this is not a likely source in PIT as the majority of macrophages when present are confined to the more luminal aspect of the plaque.
2.3 Fibroatheroma
Fibroatheroma (AHA type IV) represents a further progressive stage of atherosclerotic disease and is histologically characterized by the presence of acellular necrotic cores, which are distinct from the lipid pools of PIT as they lack expression of hyaluronan and proteoglycan versican. Recognition of early macrophage infiltration into the lipid pools and cell death along with a substantial increase in free cholesterol and breakdown of extracellular matrix, which is presumably degraded by the matrix proteases released by macrophages, is classified as “early” necrotic cores. In the early phase of the necrotic core formation an efficient system of clearance of apoptotic bodies by macrophages is present, however, the system is soon overwhelmed and there is defective phagocytic clearance of apoptotic cells and this is thought to further contribute to the vicious circle of enlargement of necrotic core and plaque progression (Fig. 2.3) [19]. As total plaque burden increases, compensatory enlargement of the vessel, i.e., positive coronary arterial remodeling, occurs to preserve arterial lumen. According to Glagov, the lumen compromise only begins to occur when the luminal narrowing exceeds >40% cross-sectional luminal narrowing [20]. We have further classified fibroatheromas into “early” and “late” based on the type of necrotic core observed. A necrotic core that is devoid of proteoglycan versican and hyaluronan or any collagen expression is termed “late” necrotic core but a thick fibrous cap fully contains the necrotic core. On the other hand “early” necrotic cores focally express the proteoglycan versican and hyaluronan, especially towards the media, but towards the lumen there is absence of matrix with macrophages infiltration.
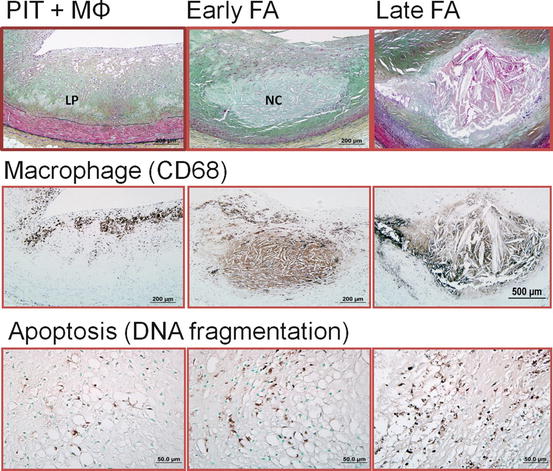
Fig. 2.3
Mechanism of necrotic core expansion in human coronary plaques. (Left column) Pathologic intimal thickening (PIT) is characterized by an underlying lipid pool (LP), which is devoid of macrophages (MΦ). The majority of macrophages when present are confined to the more luminal aspect of the plaque. Apoptotic cells are rarely seen in PIT. (Middle column) The presence of necrotic core (NC) infiltrated by CD68-positive macrophages characterizes the early stage of fibroatheroma (FA). Macrophages in early FA are capable of engulfing apoptotic bodies. (Right column) Late FA is represented by increased macrophage death and cell lysis. Free apoptotic bodies are commonly seen, possibly indicating defective clearance (efferocytosis) by resident macrophages
2.4 Intraplaque Hemorrhage
It is not only apoptotic macrophages that contribute to the accumulation of free cholesterol but other source such as red blood cells may also contribute to the expansion of the necrotic core (Fig. 2.4). Studies from our SCD registry showed that hemorrhage into a necrotic core is commonly observed in cases of plaque rupture and late necrotic core. The membranes of red blood cells are enriched with lipid, which constitutes 40% by weight, and are rich in free cholesterol content that exceeds that of all other cell membranes [21]. Excess membrane cholesterol of red blood cells can phase separate and form immiscible membrane domains consisting of pure cholesterol arranged in a tail-to-tail orientation favoring crystal formation [22]. The extent of accumulated erythrocytes incorporated into the plaque and abundant lipids, together with impaired phagocytic efficiency of macrophages to effectively clean up red blood cells and other debris, influences both the biochemical composition and size of the necrotic core [23, 24].
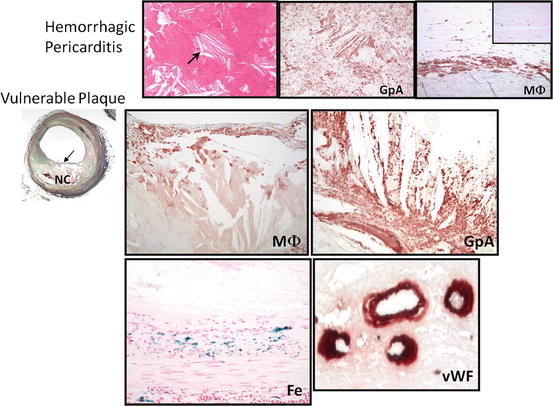
Fig. 2.4
Intraplaque hemorrhage in fibroatheroma with a late-stage necrotic core. Intraplaque hemorrhage leads to the accumulation of cholesterol monohydrate and increased lesion vulnerability. Illustrated in the top panel is the initial observation of hemorrhage in nonvascular sites showing accumulated free cholesterol. Note free cholesterol clefts in hemorrhagic pericarditis (arrow). Stained by Glycophorin A (GpA) which is specific for red blood cells, it is noted that positive staining surrounds the crystal structure of cholesterol. At the periphery of the hemorrhage, foamy macrophage (MΦ) were observed. The bottom panel shows a vulnerable plaque with macrophage positivity within the thin fibrous cap. GpA is strongly positive along with iron (Fe). Moreover, leaky microvessels within the plaque are detected as illustrated by the diffuse staining of von Willebrand factor (vWF) (reproduced with permission from Nakano et al. Vulnerable plaque. In: Zeev Vlodaver (ed) Coronary heart disease: clinical, pathological, imaging and molecular profiles. Springer)
The origin of intraplaque hemorrhage is also debatable between those claiming blood influx from luminal subsequent to plaque fissuring and proponents of leakage from intraplaque microcapillaries. However, we are in favor of the latter since often intraplaque erythrocyte extravasation is seen in the absence of plaque fissure and is associated with a high density of small vessels within the plaque. Further, some reports demonstrated the presence of incomplete mural cells coverage and dysfunctional endothelial cells of capillaries and arterioles with focal absence of basement membranes and poorly formed endothelial junctions [25]. It is possible that these immature or leaky vessels also allow diffusion of plasma proteins and diapedesis of leukocytes and erythrocytes spillage [26], which may serve as a driving force of further centripetal angiogenesis from the adventitia [27].
2.5 Hemoglobin Toxicity and Oxidative Stress
Intraplaque hemorrhage could potentially also recruit inflammatory cells [28]. The precise signaling pathways for the cellular response are not fully understood but it is postulated that hemoglobin–haptoglobin receptor CD163 on macrophages may be involved in the clearance of the complex with release of anti-inflammatory cytokines that may contribute to a decrease in inflammation [29]. However more importantly, our recent studies have elucidated the significance of oxidative stress associated with extravasated erythrocytes providing a rapid increase of hemoglobin (Hb) and continued inflammation in the intraplaque area [30]. Free Hb binds to and inactivates nitric oxide (NO), a potent molecule that plays a critical role in the regulation of smooth muscle vaso-reactivity and endothelial adhesion molecule expression, events that lead to inflammation within the vessel wall [31].
The function of haptoglobin (Hp) is primarily to handle hemoglobin released from red blood cells following intravascular or extravascular hemolysis. There are two common alleles at the Hp genetic locus denoted as 1 and 2, with two homozygous (1-1 and 2-2) and one heterozygous (2-1) genotype possible. There are functional differences between the Hp 1 and Hp 2 protein products in protecting against hemoglobin-driven oxidative stress with important functional and clinical significance. It is reported that there is a three- to fivefold increased risk of cardiovascular disease (CVD) in individuals with diabetes mellitus (DM) having the Hp 2-2 genotype as compared to DM individuals without the Hp 2-2 genotype. In particular, individuals with the Hp 2-2 genotype and diabetes mellitus appear to be at significantly higher risk of microvascular and macrovascular complications.
In atherosclerotic plaques, the primary route for clearance of the Hb–Hp complex involves the CD163 receptor expressed on immunosuppressive macrophages with M2 phenotype [32]. Physiologically low concentrations of hemoglobin (Heme) are cytoprotective as they induce the rapid upregulation of hemoxygenase-1 (HO-1). Excess pathological amounts of heme outstrip the ability of HO-1 to metabolize it so that residual heme (librating free iron) may act deleteriously on tissue by pro-oxidative and pro-inflammatory effects [33]. Also, when the capacity of protective hemoglobin-scavenging mechanisms has been saturated, levels of cell-free hemoglobin increase, resulting in the consumption of nitric oxide and resulting in clinical sequelae. NO plays a major role in vascular homeostasis and has been shown to be a critical regulator of basal and stress-mediated smooth muscle relaxation and vasomotor tone, endothelial adhesion molecule expression, and platelet activation and aggregation. Another product of excessive heme is bilirubin, which has potential antioxidant activity. Free ferrous iron has potential pro-oxidant activity, although this may be limited by its sequestration by ferritin.
2.6 Thin-Cap Fibroatheroma and Plaque Rupture
Thin-cap fibroatheroma (TCFA), traditionally designated as vulnerable plaque, is characterized by having a morphological appearance that resembles ruptured plaque [3]. TCFA generally contain a large necrotic core with overlying thin intact fibrous caps consisting mainly of type I collagen with varying degrees of macrophages and lymphocytes and very few, if any, α-actin-positive SMCs. The fibrous cap thickness is an indicator of plaque vulnerability and a TCFA is defined as having a cap thickness ≤ 65 μm since the thinnest portion of the remnant cap of a ruptured plaque was measured as 23 ± 19 μm, with 95% of ruptured caps measuring ≤ 65μm [34]. As compared to ruptured plaque, TCFA tend to have smaller necrotic cores and less macrophage infiltration. Cross-sectional luminal narrowing is also typically less in TCFA compared to ruptures and occlusive thrombus generally shows greater underlying stenosis than lesions with non-occlusive thrombus [35].
It has been shown that the site of rupture usually occurs at its weakest point, often near shoulder regions. However, in our experience, this is not always the case as we have observed an equivalent number of ruptures at the mid portion of fibrous cap, especially in individuals who are dying during exertion [36]. Therefore, it is reasonable to speculate that several processes may be involved in the mechanisms of plaque rupture, e.g., fibrous cap degradation by matrix metalloproteases (MMPs) [37], high shear stress [38], macrophage and smooth muscle cell death [39], and microcalcification and iron accumulation within the fibrous cap [40] all have been implicated.
Once plaque rupture occurs and necrotic contents are exposed to the flowing blood, this results in the triggering of the coagulation cascade in response to lipids, collagen, and tissue factors (TF). The luminal thrombus at the site of rupture is platelet rich (white thrombus) while proximal and distal to the rupture site, i.e., sites of propagation of the thrombus consist of a red thrombus composed of layers of fibrin and erythrocyte. A study of aspirated thrombi from patients presenting with AMI when examined by scanning electron microscopy (SEM) confirmed a decrease of platelet content and an increase of fibrin content as the duration of ischemia increased [41].
Related posts:
 Atherosclerosis Plaque Stress Analysis: A Review
Atherosclerosis Plaque Stress Analysis: A Review
 Quantitative Computed Tomography Analysis in the Assessment of Coronary Artery Disease
Quantitative Computed Tomography Analysis in the Assessment of Coronary Artery Disease
 Quantitative MR Analysis for the Assessment of Carotid Atherosclerosis
Quantitative MR Analysis for the Assessment of Carotid Atherosclerosis
 Carotid Angioplasty and Stenting
Carotid Angioplasty and Stenting
 Segmentation of Carotid Ultrasound Images
Segmentation of Carotid Ultrasound Images
 Carotid Plaque Stress Analysis: Issues on Patient-Specific Modeling
Carotid Plaque Stress Analysis: Issues on Patient-Specific Modeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


