Amitani and colleagues first described a unique pattern of upper lobe–predominant, idiopathic pulmonary fibrosis (IPF) in 13 patients in the Japanese literature in 1992. The currently preferred term in the English-language literature, idiopathic pleuroparenchymal fibroelastosis (iPPFE), was coined by Frankel and colleagues in 2004 in a case series of five patients presenting with unique clinical and radiographic findings distinct from any of the previously characterized idiopathic interstitial pneumonias (IIPs). Since that time, a number of cases of pleuroparenchymal fibroelastosis (PPFE) have been identified in patients with compelling etiologic correlates commonly associated with the development of interstitial lung disease (ILD), including familial interstitial pneumonitis ; recurrent lower respiratory tract lung infections ; lung, bone marrow, and hematopoietic cell transplantation ; chemotherapeutic medications (drug-induced PPFE) ; occupational exposure to aluminosilicate dust or asbestosis ; and systemic autoimmune diseases, such as rheumatoid arthritis and scleroderma. As with the other IPs/ILDs, PPFE is described as idiopathic when no associated causative etiology can be identified. iPPFE was recently recognized as a distinct entity in the American Thoracic Society/European Respiratory Society 2013 update on the IIPs and was categorized as one of the two rare IIPs, along with lymphoid IP.
Prevalence and Epidemiology
As of January 2017, approximately 120 cases have been reported overall in the published literature. To date, iPPFE appears to have no gender preference and no association with cigarette smoking. The average age at presentation is highly variable, ranging from 13 to 87 years with a median of approximately 53 years.
Clinical Presentation
Patients most often present with insidious onset of dyspnea, exercise intolerance, and nonproductive cough, similar to other patients with fibrosing ILD. Additional symptoms include chest discomfort (pain, tightness, inability to take a satisfying breath), pneumothorax, and constitutional symptoms, such as fatigue, malaise, and weight loss. Patients have been described as having a slender stature with associated “flattened thoracic cage” or “platythorax,” potentially signifying a decrease in the anterior-posterior dimension of the thorax.
Pathology
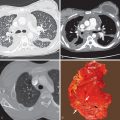
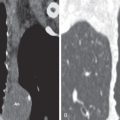
Although there are no well-defined consensus criteria regarding the histopathologic diagnosis of PPFE, in a 2013 review von der Thüsen proposed a working definition of iPPFE with “definite” and “consistent with” histologic criteria. “Definite” criteria of iPPFE include (1) upper zone fibrosis of the visceral pleura; (2) prominent, homogeneous, subpleural intraalveolar fibrosis with alveolar septal elastosis; (3) sparing of the parenchyma distant from the pleura; (4) at most, mild, patchy lymphoplasmocytic infiltrates; and (5) at most, small numbers of fibroblastic foci. Criteria that are “consistent with” iPPFE also have intraalveolar fibrosis as just described; however, (1) this may not be associated with significant pleural fibrosis, (2) may not be predominantly subpleural, or (3) may not occur in an upper lobe biopsy. Additional criteria for iPPFE were proposed by Rosenbaum and colleagues in 2015, which included (1) fibrous IP with 80% fibroelastic changes in nonatelectatic lung; (2) subpleural and/or centrilobular distribution; (3) overall inflammation absent to mild; (4) no specific lobe predilection, typically multilobar; and (5) rare or no granulomas. A recent quantitative assessment of elastic fibers (EF) demonstrated twice the number of EF in iPPFE compared with IPF. That said, it is important to note that the two conditions are not mutually exclusive and often appear to coexist. Although an experienced pathologist may be able to recognize the characteristic features of PPFE on standard eosin and hematoxylin stains, incorporating the routine use of EF-specific agents, such as orcein or Verhoeff–van Gieson stains, could greatly facilitate identifying PPFE histologically ( Fig. 36.1 ).

Physiology
The main physiologic abnormality is a restrictive ventilatory impairment similar to other fibrosing ILDs. Often there is a disproportionate reduction in forced vital capacity relative to the reduction seen in the diffusion capacity of carbon monoxide compared with the degree of impairment seen in these parameters in patients with other ILDs, such as IPF. Additionally, disproportionate upper lobe fibrotic collapse with compensatory hyperinflation of the lower lobes may increase the ratio of residual volume to total lung capacity distinguishing PPFE from IPF.
Imaging
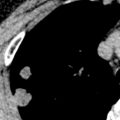
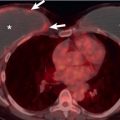
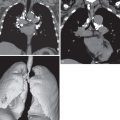
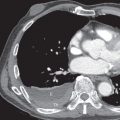
Radiographs in early iPPFE may demonstrate bilateral apical pleural irregularity or be normal as subtle visceral pleural nodularity, and intraalveolar fibrosis may be radiographically occult ( Fig. 36.2 ). Indeed, patients often present with physiologic impairment and symptoms that are more pronounced than what would be expected based upon the radiographic findings alone. Later-stage disease classically demonstrates increased severity in the apical pleural thickening, with upper lobe–predominant reticulonodular opacities and retracted, elevated hila. Lateral radiographs may illustrate anterior-posterior dimensional narrowing. Recurrent pneumothorax may occur in PPFE.