Endocrine disorders associated with adrenal pathologies can be caused by insufficient adrenal gland function or excess hormone secretion. Excess hormone secretion may result from adrenal hyperplasia or hormone-secreting (ie, functioning) adrenal masses. Based on the hormone type, functioning adrenal masses can be classified as cortisol-producing tumors, aldosterone producing tumors, and androgen-producing tumors, which originate in the adrenal cortex, as well as catecholamine-producing pheochromocytomas, which originate in the medulla. Nonfunctioning lesions can cause adrenal gland enlargement without causing hormonal imbalance. Evaluation of adrenal-related endocrine disorders requires clinical and biochemical workup associated with imaging evaluation to reach a diagnosis and guide management.
Key points
- •
Endocrine disorders associated with adrenal pathologies can be caused by insufficient adrenal gland function or excess hormone secretion.
- •
Excess hormone secretion may result from adrenal hyperplasia or hormone-secreting (ie, functioning) adrenal masses.
- •
Based on the hormone type, functioning adrenal masses can be classified as cortisol-producing tumors, aldosterone producing tumors, and androgen-producing tumors, which originate in the adrenal cortex, as well as catecholamine-producing pheochromocytomas, which originate in the medulla.
- •
Nonfunctioning lesions can cause adrenal gland enlargement without causing hormonal imbalance.
- •
Evaluation of adrenal-related endocrine disorders requires clinical and biochemical workup followed by imaging evaluation to reach a diagnosis and guide management.
Introduction
A wide spectrum of pathologies can affect the adrenal gland, including a range of functioning benign and malignant masses that secrete increased amounts of adrenal hormones as well as nonfunctioning tumors that do not affect hormone levels. Functioning tumors or hyperplasia of the adrenal cortex can cause Cushing syndrome (CS) from cortisol excess, Conn syndrome from aldosterone overproduction, or hyperandrogenism from androgen excess. Pheochromocytomas are tumors that arise from the adrenal medulla and secrete catecholamines. The other well-known adrenal pathology is adrenal insufficiency, which is characterized by low levels of adrenal hormones, and may result from adrenalectomy, autoimmune disorders, granulomatous diseases, hemorrhage, bilateral adrenal metastases, or infections causing adrenal gland destruction and hypofunction. The diagnosis of adrenal-related endocrine disorders is usually based on physical examination and biochemical workup, followed by imaging evaluation. Proper management of endocrine disorders, especially those that are neoplastic, requires a multidisciplinary team collaboration among endocrinologists, radiologists, pathologists, and surgeons. In this review, we focus on adrenal disorders associated with an endocrine abnormality and will not include nonfunctioning adrenal tumors.
Imaging evaluation
Imaging evaluation guides the appropriate management of adrenal-related endocrine disorders. Computed tomography (CT) with adrenal mass–dedicated protocol is the reference standard imaging modality for assessment of adrenal gland pathologies. An adrenal CT is performed with 2.5 to 3 mm thin slices, before and after intravenous administration of 100 to 150 mL of iodinated contrast material. The adrenal mass protocol includes density measurement of the mass on noncontrast CT. An unenhanced attenuation value less than 10 Hounsfield units (HU) is characteristic of a lipid-rich adenoma. However, for adrenal masses with attenuation values greater than 10 HU, the absolute percentage of enhancement washout is calculated by measuring the unenhanced attenuation, the enhanced attenuation at 60 seconds, and attenuation 15 minutes after contrast injection to show rapid enhancement and rapid washout.
CT is often the first-line modality for adult adrenal imaging due to its wide availability, reproducibility, good temporal resolution and better spatial resolution than MR imaging. Adrenal abnormalities can be detected and characterized using contrast-enhanced CT with or without noncontrast images, as mentioned previously.
Other imaging modalities, such as MR imaging and PET with fluorodeoxyglucose (FDG) or other specialized radioactive tracers such as gallium-68 DOTATATE are useful as adjuncts to CT when CT is negative or inconclusive. Ultrasonography can be considered as an imaging modality for adrenal lesions in neonates and young children ; however, because the echogenicity of adrenal glands is close to the echogenicity of retroperitoneal fat, it is a challenge to visualize adrenal glands on ultrasound in older children and adults. In addition, ultrasound has limitations in identifying small lesions, and overlying bowel gas can be an impediment to adequate visualization.
MR imaging, which is discussed in greater detail later in this article, is often used as problem solver, especially when CT is contraindicated or inconclusive. MR imaging has a particular advantage of the lack of ionizing radiation. Chemical shift in-phase and opposed-phase are the most important MR imaging pulse sequences for characterizing adrenal masses by detecting intracytoplasmic lipid, high level of which is most often indicative of benign adrenal adenoma, with some exceptions such as lipid-containing metastases, collision tumors, or adrenocortical cancers. Ultimately, CT has been found to have the upper hand, compared with MR imaging, for differentiating between lipid-poor adrenal adenomas, which show characteristic delayed postcontrast washout without detectability of lipid on chemical shift MR imaging, and other indeterminate adrenal masses. T2-weighted MR imaging may be helpful in the diagnosis of pheochromocytoma, which demonstrates marked hyperintensity; also known as “lightbulb sign.”
Nuclear medicine imaging modalities such as MIBG and indium-111 octreotide and gallium-68 DOTATATE PET are commonly used to evaluate pheochromocytoma. FDG-PET shows promise in the differentiation of malignant cortical lesions from benign masses. Malignant lesions show increased FDG uptake, and FDG-PET has been shown to have 94% to 100% sensitivity and 80% to 100% specificity in the detection of malignant masses.
Overview of adrenal-related endocrine disorders
Endocrine disorders associated with adrenal pathologies may be subclinical or clinical and can be classified into hypofunctioning disorders, such as adrenal insufficiency, or hyperfunctioning disorders, such as hyperaldosteronism, hypercortisolism, catecholamine excess, and hyperandrogenism.
Adrenal Insufficiency
Primary adrenal insufficiency occurs due to partial or complete destruction of the adrenal cortex, and patients with this condition may present with hyponatremia, hyperkalemia, fatigue, muscle weakness, hypotension, weight loss, abdominal pain, and hyperpigmentation. Common etiologies of primary adrenal insufficiency are autoimmune disease (ie, Addison disease), granulomatous diseases such as tuberculosis, other infections, neoplasms such as metastases and lymphoma, adrenal gland hemorrhage, adrenoleukodystrophy, and congenital adrenal hyperplasia.
Screening tests for adrenal insufficiency include morning cortisol and adrenocorticotropic hormone (ACTH) levels or a high-dose cosyntropin (synthetic ACTH) stimulation test. Cortisol levels that fail to rise after ACTH stimulation and remain lower than 20 μg/dL indicate adrenal insufficiency. , In primary adrenal insufficiency, the synthesis and release of adrenocortical hormones are impaired. The next step after the diagnosis of primary adrenal insufficiency is to check adrenal autoantibodies for autoimmune adrenal insufficiency. If autoantibodies are negative, very long chain fatty acids can be tested for adrenoleukodystrophy, serum 17-hydroxyprogesterone level can be measured for congenital adrenal hyperplasia, and CT can be performed to detect adrenal hemorrhage, infection, infiltration, or malignancy. In adrenoleukodystrophy, very long chain fatty acids are accumulated in tissue and body fluids, and the clinical manifestations can be seen as central nervous system demyelination and primary adrenal insufficiency. Adrenal insufficiency is usually associated with cerebral adrenoleukodystrophy or adrenomyeloneuropathy involving the spinal cord and peripheral nerves, but in 8% of cases adrenal insufficiency is the only clinical manifestation. Loes and colleagues described 5 different MR imaging patterns of cerebral adrenoleukodystrophy, and the radiologic finding indicative of adrenal involvement is adrenal cortex atrophy. In congenital adrenal hyperplasia, bilateral enlarged adrenal glands, wrinkled surface, and cerebriform pattern are characteristic ultrasound findings; however, the adrenal glands may also appear normal. Therefore, ultrasonography should only be used as an adjunct to CT or MR imaging when adrenal hyperplasia is suspected.
CT imaging findings of primary adrenal insufficiency depend on the etiology and course of the disease. For instance, adrenal hemorrhage can present as a high-attenuation mass in the acute stage, then decrease in attenuation with aging of the hematoma.
Infectious diseases and autoimmune and granulomatous disorders can cause bilateral enlargement of the glands in the acute/subacute stage, with atrophy and often calcification of adrenal glands in the chronic stage. For example, Addison disease is considered subacute when present for less than 2 years, and in these cases, CT usually shows bilateral enlarged adrenal glands with occasional central necrosis and peripheral rim enhancement due to adrenalitis. ,
In the chronic stage of adrenal insufficiency, CT shows bilateral adrenal gland atrophy. Calcifications are seen in 50% of cases of primary adrenal insufficiency secondary to tuberculosis; however, calcifications due to tuberculosis cannot be distinguished radiologically from other causes of calcifications, such as prior hemorrhage or idiopathic calcifications. Clinical and hormonal correlation is the key in such cases.
Adrenal hemorrhage can result from blunt trauma, anticoagulation therapy, sepsis, or stress following surgery or severe burns, or it can be a complication of adrenal venous sampling. In the setting of trauma, adrenal hemorrhage usually occurs in trauma of higher severity, and isolated adrenal hemorrhage is rare. Hemorrhage can also be seen in both benign and malignant masses such as adenomas, adrenal cortical carcinomas, myelolipomas, and pheochromocytomas. Bilateral involvement can be seen in 20% of adrenal hemorrhage cases. The most common imaging feature of hemorrhage, a high-attenuating adrenal mass, is seen in 83% of cases on unenhanced CT ( Fig. 1 ). On MR imaging, adrenal hemorrhage shows intermediate signal intensity on T1-weighted images and low signal intensity on T2-weighted images in the acute stage and high signal intensity on both T1-weighted and T2-weighted images in the subacute stage. The typical appearance of chronic adrenal hemorrhage includes high signal intensity on T1-weighted images and hypointense peripheral rim on T2-weighted images. The high signal intensity on T1-weighted images can be due to methemoglobin, and hypointense peripheral rim on T2-weighted images is seen due to hemosiderin. Hematomas gradually decrease in size and may completely resolve or develop calcifications within a few months.

Granulomatous infections such as tuberculosis or histoplasmosis account for 10% to 30% of primary adrenal insufficiency cases. CT findings in the acute stage are bilateral adrenal enlargement, central necrosis, and peripheral rim enhancement. In the subacute and chronic stages, the adrenal glands become atrophic and calcified.
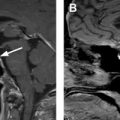
Other causes of adrenal insufficiency include lymphoma ( Fig. 2 ) and metastases ( Fig. 3 ). Lymphomatous adrenal lesions are usually homogeneous and large, but they can also be heterogeneous due to necrosis or hemorrhage. Adrenal lymphoma is bilateral in 50% of cases and is usually accompanied by retroperitoneal lymphadenopathy. On CT, adrenal lymphoma usually demonstrates soft tissue attenuation and shows mild progressive enhancement. On MR imaging, adrenal lymphoma typically exhibits low signal intensity on T1-weighted images and high signal intensity on T2-weighted images, with mild to moderate enhancement.


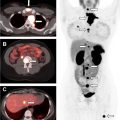
The adrenal glands are a common site for hematogenous metastases because of their abundant blood supply, and most patients with adrenal metastases have a history of cancer. In a study by Song and colleagues, not a single malignant lesion was found in 1049 patients without a history of malignancy who had adrenal nodules incidentally discovered at imaging. It is also rare to find an incidental adrenal metastasis at the initial presentation of an unknown primary malignancy. In a series of 1639 patients with unknown primary malignancy, the adrenal glands were involved in only 6%, and involvement was limited to the adrenal glands in only 0.2%; furthermore, all of the adrenal lesions were 6 cm or larger, and 75% of the patients had bilateral involvement. Metastases to the adrenal glands most commonly originate from lung, breast, liver, kidney, and gastric cancers. On CT, adrenal metastases usually have attenuation values greater than 10 HU on unenhanced series, and they show irregular peripheral or heterogeneous enhancement on contrast-enhanced series. Unlike adenomas, metastases enhance rapidly but do not exhibit enhancement washout, with the exception of some hypervascular metastatic lesions from primary tumors such as renal cell carcinoma and hepatocellular carcinoma, which can show rapid washout and hence can be misdiagnosed as adenomas. Similarly, the presence of intracellular fat is highly specific for benign adenoma; however, some metastases, such as those from renal cell or hepatocellular carcinoma, can rarely contain intracellular lipid and mimic adrenal adenomas. Associated hemorrhage and calcifications have been also described within these lesions. On MR imaging, metastatic masses typically demonstrate high signal intensity on T2-weighted images and low signal intensity on T1-weighted images and with heterogeneous postcontrast enhancement.
Hyperaldosteronism
Hyperaldosteronism can be primary, also known as Conn syndrome (the classic description of which is that of an aldosterone-producing adenoma), or secondary depending on the renin level. Whereas aldosterone levels in both primary and secondary hyperaldosteronism are high, the renin level is low in primary and high in secondary hyperaldosteronism (such as seen with a renin-secreting tumor or renovascular hypertension). Hyperaldosteronism presents with hypertension (typically refractory to multidrug therapy), hypokalemia (which is not always present), metabolic alkalosis, muscle cramps/weakness, headache, increased thirst, and frequent urination.
Common etiologies of secondary hyperaldosteronism are congestive heart failure, cirrhosis, nephrotic syndrome, renal artery stenosis, renin-secreting tumors, or diuretic use. Adrenal lesions in primary hyperaldosteronism can be unilateral or bilateral adrenal adenoma(s) ( Fig. 4 ), adrenal hyperplasia, or adrenal carcinoma. Conn syndrome is caused by an aldosterone-producing adenoma in 35% of cases and bilateral idiopathic hyperplasia in 60% of cases. ,

Screening tests for hyperaldosteronism include measuring the plasma renin activity (PRA) and plasma aldosterone concentration (PAC). Primary aldosteronism is suggested if the PAC:PRA ratio is greater than 20, and PAC levels greater than 20 ng/dL and PRA less than1 ng/mL per hour in the setting of spontaneous hypokalemia confirm the diagnosis. If both PRA and PAC are elevated, etiologies for secondary hyperaldosteronism should be further investigated. On the other hand, decreased PRA and PAC levels should prompt an investigation of other etiologies such as exogenous mineralocorticoids (eg, licorice ingestion) and CS.
For confirmation of primary aldosteronism, measuring 24-hour aldosterone levels after oral sodium loading, measuring aldosterone response to intravenous saline infusion, and fludrocortisone suppression and captopril challenge tests may be used. Aldosterone level higher than 10 ng/dL in a saline infusion test and direct renin concentration lower than 24 mIU/mL and PRA lower than 2 ng/mL hour in fludrocortisone suppression test are confirmatory results.
After the biochemical confirmation of primary hyperaldosteronism, adrenal imaging should be pursued. If the patient has a unilateral adrenal adenoma on imaging and is younger than 40 years, unilateral adrenalectomy is the recommended treatment because the likelihood of this representing an incidental nonfunctional adrenal adenoma is only 1% to 2%. Patients older than 40 years with a unilateral adrenal adenoma and patients with normal glands or equivocal findings should be further investigated with selective adrenal venous sampling, which is the reference standard test for differentiating unilateral from bilateral adrenal disease. A coincidental nonfunctioning neoplasm should be considered if adenomas are larger than 2 cm, because aldosteronomas are usually smaller than 1.5 cm. If imaging shows bilateral adrenal gland thickening or micronodular changes, the diagnosis is bilateral adrenal hyperplasia or idiopathic hyperaldosteronism. If adrenal venous sampling confirms bilateral aldosterone production, medical management with an aldosterone receptor antagonist should be undertaken. In addition, glucocorticoid-remediable aldosteronism, wherein aldosterone secretion is under the control of ACTH, also presents with nonlocalizing venous sampling test results.
In this setting, adenomas can be detected on CT in only 70% of hyperaldosteronism cases. Aldosteronomas are typically homogeneous hypoattenuating nodules with attenuation less than 10 HU (if lipid-rich) and homogeneous contrast enhancement on CT, and they rarely show calcifications. Because the average diameter of aldosteronomas is less than 2 cm (approximately 20% measure <1 cm), CT should be performed with a slice thickness of 5 mm or less. On MR imaging, they are iso- or hypointense relative to the liver on T1-weighted images and slightly hyperintense on T2-weighted images, with decrease in signal intensity on opposed-phase compared with in-phase pulse sequences. It has been reported that aldosteronomas have the lowest attenuation values among hyperfunctioning adrenal adenomas. , As many as 50% of surgically proven aldosteronomas may be misdiagnosed as hyperplasia on CT. Adrenal cortical hyperplasia is among the etiologies of patients presenting with excessive secretion of adrenal cortex hormones such as cortisol, aldosterone, and androgens, and 60% of hyperaldosteronism cases result from adrenal cortical hyperplasia. Adrenal cortical hyperplasia typically presents as smooth to slightly lobular, uniform enlargement, although macronodular morphology can be also seen. In diffuse hyperplasia, there is homogeneous enlargement of the entire gland, and the normal inverted V or Y configuration is maintained. The limbs of the adrenal glands usually measure more than 5 cm in length and more than 1 cm in thickness. The attenuation and signal intensity of cortical adrenal hyperplasia are usually similar to that of the normal gland ( Fig. 5 ). In a small percentage of patients, low CT attenuation and signal drop on opposed-phased MR imaging can be seen, especially in cases with macronodules. , ,

Hypercortisolism
The chronic excess of cortisol causes CS, which presents with features that include obesity, moon facies, proximal muscle weakness, thin skin with easy bruising, violaceous striae, abnormal body fat distribution (ie, truncal obesity, fullness of the dorsal and supraclavicular fat pads), acne, hirsutism, hypertension, edema, low bone mass and fractures, amenorrhea, and impaired glucose tolerance. CS arises from exogenous glucocorticoid administration or endogenous overproduction of cortisol, which is either mediated by ACTH (ACTH-dependent CS) or caused by the neoplastic hypersecretion of cortisol from the adrenal cortex (ACTH-independent CS). ACTH-dependent hypercortisolism accounts for 80% of cases and is typically caused by an ACTH-producing pituitary tumor (Cushing disease) or more rarely by an ectopic ACTH-producing neuroendocrine tumor (NET) or a corticotropin-releasing hormone (CRH) producing tumor. The most likely source of ectopic ACTH production is a thoracic tumor (eg, bronchopulmonary NET, small cell lung cancer, thymic NET), but ectopic ACTH secretion can also occur in pancreatic NETs and, more rarely, pheochromocytomas and medullary thyroid carcinomas. CRH-producing tumors account for fewer than 1% of the cases. ACTH-dependent CS can present with bilateral diffuse and uniform adrenal enlargement (see Fig. 5 ), which in time becomes nodular in appearance, and this is known as multinodular hyperplasia. Occasionally these ACTH-dependent macronodules grow larger and become autonomous, an entity called massive macronodular hyperplasia. , ACTH-independent CS can result from an adrenal adenoma, carcinoma, primary pigmented nodular adrenocortical disease (PPNAD) or ACTH-independent macronodular adrenocortical hyperplasia ( Fig. 6 ), also known as bilateral macronodular adrenal hyperplasia. Adenomas account for 60% of adrenal-related CS cases, and the remaining 40% adrenal-related CS cases are caused by adrenocortical carcinoma.