Tracer
[11C]GSK931145
[18F]MK-6577
[11C]CMPyPB
[11C]RO501852
[11C]RO5013853
[11C]-N-Methyl_SSR504734 ([11C]GSK565710)
Structure
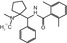
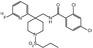
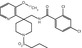
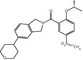
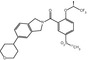
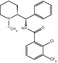
MW
350.51
487.09
499.11
443.19
497.16
410.87
LogD (pH7.4)
2.5
2.9
3.2
2.2
2.3
2.97
pIC50(GlyT1)
8.4
8.7 (human)
8.4 (human)
NR
NR
8.3
pIC50(GlyT2)
<4.6
<5
NR
NR
NR
5.4
Affinity GlyT1
pKi = 8.97 (rat cortex)
pKi = 8.94 (human)
NR
K D = 3.9(rat); K D = 3.7(human)
K D = 2(rat); KD = 1.7(human)
pKi = 9.09 (rat)
f P (%)
0.8 (human)
NR
NR
NR
NR
NR
Primate
Yes
Yes
Yes
Yes
Yes
Yes
Human
Yes
Yes
No
No
Yes
No
%COV(V T)
29–38 % (human)
–
–
12 % (human)
–
12.4.1 [11C]ALX5407
The first GlyT1 inhibitor to be labelled with a PET isotope was [11C]ALX5407 (Ravert et al. 2001), but no information on in vitro or in vivo evaluation has been provided subsequently, suggesting that this tracer was not successful as a PET ligand.
12.4.2 [11C]GSK931145
In 2008, the imaging group at GSK reported the evaluation of three candidate PET ligands, [11C]GSK931145, [11C]GSK565710 and [11C]GSK991022, in porcine CNS at the Society of Nuclear Medicine Annual Meeting in New Orleans (Passchier et al. 2008). Full data and additional results were published in 2010 (Passchier et al. 2010) and confirmed that out of the three labelled compounds, [11C]GSK931145 showed promise as a potential PET ligand. Regional estimates of the total volume of distribution reflected the known rank order of GlyT1 distribution for [11C]GSK931145 (brainstem > cortex) and were reduced to a homogeneous level following pre-administration of 0.5 mg/kg GSK565710. Calculation of binding potential (BPND), under the assumption of full block with 0.5 mg/kg GSK565710, indicated a BPND ~ 2 in the brainstem. Subsequent studies in nonhuman primates and human subjects confirmed suitable dosimetry for multiple scans in human subjects with a mean effective dose in humans of 4.02 μSv/MBq (male phantom) and 4.95 μSv/MBq (female phantom). Little or no urinary excretion was observed and the limiting organ was the liver with radiation absorbed dose estimates of 9.89 μGy/MBq and 13.05 μGy/MBq for male and female phantoms, respectively (Bullich et al. 2011). Further studies in healthy volunteers demonstrated the lack of a reference region devoid of specific binding. Test-retest variability of regional total volume of distribution (V T ) was poor (>30 %), and overall signal to noise was much reduced in man compared to pig and primate. More detailed analysis pointed towards differences in plasma protein binding as the leading cause for the observed species differences. Although [11C]GSK931145 does not appear suitable to investigate population differences (unless they are large), it can be used to determine occupancy and derive an EC50 value using a pseudo reference region approach (Gunn et al. 2011a), as demonstrated for the GlyT1 drug candidate GSK1018921.
12.4.3 [18F]MK-6577
Hamill et al. (2011) reported the preclinical evaluation of [18F]MK-6577 and [11C]CMPyPB in rhesus monkey as potential PET ligands for delineation of GlyT1 in the CNS. Both tracers showed good brain uptake with a distribution that was in agreement with reported rank order of GlyT1 and with results obtained with [11C]GSK931145 in primate brain. Although initial results looked promising, no further work was reported for [11C]CMPyPB. Selectivity of MK-6577 for GlyT1 over GlyT2 was established by measuring inhibition of [14C]glycine uptake in HEK cells expressing human GlyT2. A similar experiment was performed to determine selectivity for GlyT1 over the taurine transporter TauT in JAR cells. In addition, no off-target activities were observed when MK-6577 was screened against >100 known cerebral receptors, ion channels and enzymes. Blocking studies with (S)-CPyPBE resulted in rapid washout of [18F]MK-6577 from all brain regions with near homogeneous tracer uptake throughout the brain. As observed for [11C]GSK931145, reduction was seen for all brain regions. A subsequent chase study using (S)-CPyPBE demonstrated reversibility of [18F]MK-6577 binding. Further papers have focused on quantification of GlyT1 availability using a model-based input function (Sanabria-Bohórquez et al. 2012), comparison of [18F]MK6577 and [11C]GSK931145 scan results in baboons (Zheng et al. 2013) and the use of [18F]MK-6577 to estimate glycine transporter occupancy of drug candidates (Eddins et al. 2013), but to date no human data has been reported.
12.4.4 [11C]Ro5013853
The radiosynthesis of another PET probe for GlyT1, [11C]Ro5013853, was also reported in 2011 (Pinard et al. 2011). Recently, Borroni et al. (2013) and Wong et al. (2013) reported the preclinical and clinical characterization of this radiopharmaceutical as a novel PET tracer for GlyT1. Wong et al. (2012) and Borroni et al. (2013) also evaluated [11C]Ro5013852, but although this compound did show a distribution in agreement with the previous PET ligands, [11C]Ro5013853 displayed slightly higher brain uptake and a higher specific signal as determined using the occipital cortex as reference region. Thus, [11C]Ro5013853 was selected for further studies.
The reported intra-subject variability (%COV) for V T in baboon was good across brain regions, ranging from 18 % for pons to 10 % for cerebellum, thalamus and cortex. The characterization study in humans demonstrated that similar to [11C]GSK931145, regional brain V T for [11C]RO5013853 was substantially reduced in man compared to baboon. The BPND estimates in cerebellum, pons and thalamus were comparable to those obtained for [11C]GSK931145; however, [11C]RO5013853 test-retest variability was superior and can be smaller than 10 % (Wong et al. 2013). Although the authors claim that both [11C]RO5013852 and [11C]RO5013853 are selective for GlyT1 in a screen of more than 80 receptors, ion channels and enzymes widely distributed in the brain, no information of selectivity for GlyT1 over the anticipated closest pharmacology, GlyT2 was provided.
[11C]RO5013853 was successfully applied to measuring the GlyT1 occupancy as a result of oral dosing of bitopertin in baboon and subsequently in man (Martin-Facklam et al. 2013).
12.4.5 [11C]-N-Methyl-SSR504734
Most recently, Fuchigami et al. (2012) reported results in primate CNS for [11C]-N-Methyl-SSR504734. This tracer was previously reported by Passchier et al. (2010) as [11C]GSK565710 and evaluated in porcine brain where it was found to be inferior to [11C]GSK931145. In cynomolgus monkey, the authors found [11C]-N-Methyl-SSR504734 to be a promising potential GlyT1 PET ligand. Baseline V T for the two monkeys followed the known rank order for GlyT1 distribution: thalamus > pons > cerebellum = putamen > cortical regions > white matter. Volumes of distribution for monkey 1 were ~ 2-fold higher than observed for monkey 2. Specific binding was characterised through pretreatment with SSR504734, leading to a reduction in V T compared to baseline and an apparent occupancy of 45 and 74 % for doses of 1.5 and 4.5 mg/kg, respectively. Interestingly, thalamus seemed to behave differently from other regions which the authors propose may be due to a different non-displaceable component or a different GlyT1 occupancy. Further work using structurally different blocking agents will be required to confirm this finding. The authors plan to investigate the behaviour of this potential PET ligand in human volunteers.
Related posts:
 The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
The Impact of Genetic Polymorphisms on Neuroreceptor Imaging
 Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
Imaging of Central Benzodiazepine Receptors in Chronic Cerebral Ischemia
 PET Tracers for Beta-Amyloid and Other Proteinopathies
PET Tracers for Beta-Amyloid and Other Proteinopathies
 PET Imaging of ABC Transporters in the BBB
PET Imaging of ABC Transporters in the BBB
 PET and SPECT Imaging of Steroid Hormone Receptors
PET and SPECT Imaging of Steroid Hormone Receptors
 Imaging Histamine Receptors Using PET and SPECT
Imaging Histamine Receptors Using PET and SPECT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


