Introduction
Interstitial lung disease (ILD) refers to a broad category of diffuse parenchymal lung diseases characterized by inflammation and/or fibrosis of the lungs. Patients invariably present with dyspnea of varying time course and severity. Patients are often hypoxemic, and pulmonary function tests typically demonstrate a restrictive pattern, with reduced diffusing capacity. The term interstitial lung disease is somewhat of a misnomer because many ILDs also involve the alveolar spaces. To date, more than 150 ILDs have been described. Imaging, particularly high-resolution computed tomography (HRCT), plays a pivotal role in the evaluation, diagnosis, and monitoring of ILD. In this chapter, we begin by discussing the imaging modalities and techniques used to evaluate ILD. We then describe the idiopathic interstitial pneumonias (IIPs), which are a subset of ILDs of unknown cause but with distinct clinicopathologic descriptions. Of the various IIPs, we spend the most time discussing usual interstitial pneumonia because it is the most common IIP and has the most detailed diagnostic criteria. Other common ILDs, including sarcoidosis, hypersensitivity pneumonitis (HP), pneumoconioses, and ILDs associated with collagen vascular diseases, are discussed elsewhere in this text. Finally, we conclude with a brief summary of emerging knowledge on the genetics of pulmonary fibrosis.
Appropriateness of Imaging Modalities
Appropriate Modalities in Suspected Interstitial Lung Disease
Chest radiography is one of the initial screening tests for ILD due to its wide availability, low cost, and low radiation exposure to the patient. Posteroanterior and lateral chest radiographs are commonly ordered for patients with dyspnea. However, early manifestations of ILD are difficult to perceive on chest radiographs. In fact, chest radiographs are normal in up to 15% of patients with ILD. And, even in cases of advanced ILD, it can be challenging to characterize radiographic findings and formulate a differential diagnosis.
CT is the imaging modality of choice for the evaluation of ILD. More specifically, HRCT, in which images are reconstructed with thin sections and high spatial frequency algorithms, is recommended to image the lung interstitium optimally and characterize parenchymal abnormalities. Since its introduction over 30 years ago, HRCT has emerged as an indispensable diagnostic tool in the evaluation of patients with suspected ILD.
MRI has limited utility in the evaluation of ILD. MRI suboptimally images the lungs due to the inherent absence of proton density in the aerated lungs, as well as increased susceptibility artifact from extensive air–soft tissue interfaces. However, due to the concern of cumulative radiation exposure in patients with chronic pulmonary disease, MRI has been used in lieu of CT at some centers to monitor certain patient populations. For example, MRI surveillance of patients with cystic fibrosis has been well reported in the literature.
Standard CT Protocol for Interstitial Lung Disease Assessment
The standard HRCT protocol for ILD assessment includes an inspiratory scan with the patient in the supine position, an inspiratory scan in the prone position, and an expiratory scan in the supine position. In the current multidetector CT (MDCT) era, these scans are typically performed helically, with whole-lung volumetric acquisition. However, the prone and expiratory scans may be performed with individual axial scans at spaced (1–4 cm) intervals if radiation exposure is a concern. The HRCT protocol may be tailored to the clinical indication. For example, the prone scan may be omitted in patients without suspected ILD or with advanced lung disease. However, in a busy clinical practice, these modifications to the HRCT protocol may prove to be challenging.
Rationale for Including Prone Images
The supine inspiratory HRCT is adequate for diagnosis in most cases. However, prone images can be valuable in detecting subtle or early ILD. Subsegmental atelectasis is often present in the dependent lungs in normal individuals, appearing as dependent subpleural densities or lines. These findings, unfortunately, can mimic those of early lung ILD. However, on prone scanning, the posterior subsegmental atelectasis resolves due to increased posterior lung aeration, whereas true subpleural disease remains visible ( Fig. 19.1 ). As mentioned previously, in patients with advanced ILD, prone images may be omitted because subtle dependent opacities no longer pose a diagnostic dilemma.


Rationale for Including Expiratory Images
Expiratory scanning is a useful adjunct to the inspiratory scan in the evaluation of patients with suspected small airways or obstructive lung disease. Air trapping is diagnosed by identifying areas of relative lucency on expiratory images that maintain the same attenuation as on the corresponding inspiratory images ( Fig. 19.2 ). Air trapping on expiratory HRCT has been shown to correlate with obstructive deficits on pulmonary function testing. Although air trapping is not a prominent component of most ILDs, the presence of air trapping may occasionally aid in the differential diagnosis. Moreover, the cause of the patient’s symptoms may be due to obstructive lung disease rather than restrictive ILD. Consequently, we recommend that expiratory scans be routinely acquired in every patient’s initial HRCT assessment.


Ensuring That CT Images Were Acquired in Expiration
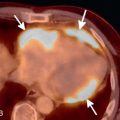
Expected CT findings during expiration include an increase in lung attenuation, decrease in cross-sectional lung area, and reduction in airway size. Of these, reduction in airway size, particularly the trachea, is most useful in determining if the patient performed an adequate expiratory maneuver. During inspiration, the trachea maintains a round or elliptic shape. During expiration, the trachea assumes more of a crescent shape as the membranous posterior wall bows anteriorly ( Fig. 19.3 ). The mean cross-sectional area of the trachea can decrease by up to half of its area on inspiration. If the tracheal morphology and area do not change between inspiratory and expiratory scans, the patient may not have reached an adequate level of expiration. On the other hand, obliteration of the tracheal lumen during expiration is diagnostic of flaccidity of the supporting tracheal cartilage, known as tracheomalacia.


Usual Interstitial Pneumonia
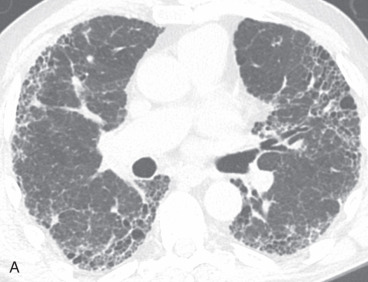
Usual interstitial pneumonia (UIP) is a chronic fibrosing interstitial pneumonia in which there is a spatially and temporally heterogeneous distribution of normal lung, interstitial inflammation, fibrosis, and honeycomb change ( Fig. 19.4 ). A characteristic histologic lesion is the fibroblastic focus, which represents an aggregate of proliferating fibroblasts and myofibroblasts. UIP represents the histopathologic pattern associated with idiopathic pulmonary fibrosis (IPF). UIP/IPF is the most common IIP, accounting for 50% to 60% of cases. It is important to make the diagnosis of UIP because IPF has a poor prognosis, with a median survival of less than 5 years after the time of diagnosis. Unlike other ILDs, IPF does not respond to conventional corticosteroid and/or immunomodulator therapy. Moreover, given recent US Food and Drug Administration (FDA) approval of medications shown to slow functional decline in IPF patients, accurate diagnosis is paramount.

Clinical Causes
The differential diagnosis for UIP consists of IPF (majority of cases in most clinics), connective tissue diseases, drug toxicity, chronic HP, and pneumoconioses. Although IPF is the most common cause of UIP, it is a diagnosis of exclusion and should only be made when other possible causes of UIP and ILD have been excluded.
Classification System on HRCT
In 2011 the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Association published an evidence-based consensus statement on the diagnosis and management of IPF. In 2018, the same organizations published revisions of the original recommendations. The updated document established four categories of HRCT patterns in patients with suspected IPF: (1) UIP; (2) probably UIP; (3) indeterminate for UIP; and (4) alternative diagnosis. The radiologist reading these scans should make every effort to classify the HRCT findings as one of these four patterns.
Establishing Definite Diagnosis on HRCT
According to the 2018 updated guidelines, the features that are required to make a diagnosis of a UIP pattern on HRCT are (1) subpleural and basal predominant; distribution is often heterogeneous; and (2) honeycombing with or without peripheral traction bronchiectasis or bronchiolectasis ( Table 19.1 ). A number of studies have shown that a UIP pattern on HRCT is highly predictive of a histopathologic UIP pattern. In patients with suspected IPF who do not have identifiable causes of ILD, fulfillment of these HRCT criteria is diagnostic of IPF and obviates the need for surgical lung biopsy.
| UIP PATTERN | PROBABLE UIP PATTERN | INDETERMINATE FOR UIP PATTERN | ALTERNATIVE DIAGNOSIS PATTERN |
|---|---|---|---|
| Subpleural and basal predominant; heterogeneous distribution | Subpleural and basal predominant; heterogeneous distribution | Subpleural and basal predominant | CT features: cysts, marked mosaic attenuation, predominant GGO, profuse micronodules, centrilobular nodules, nodules, consolidation |
| Honeycombing with or without traction bronchiectasis/ bronchiolectasis | Reticular pattern with traction bronchiectasis/ bronchiolectasis | Subtle reticulation; may have mild GGO or distortion | Distribution: peribronchovascular, perilymphatic, upper or mid-lung |
| May have mild GGO | CT features or distribution of fibrosis that do not suggest any specific etiology | Other: pleural plaques, dilated esophagus, distal clavicular erosions, extensive lymph node enlargement, pleural effusions or thickening |
Importance of Honeycombing
Honeycombing is critical to make a definitive diagnosis of a UIP pattern on HRCT. Honeycombing appears as clustered cystic air spaces with well-defined walls, typically subpleural in location. If honeycombing is absent but other features of UIP are present, the HRCT findings are best classified as a “probable UIP” pattern. In a patient with high pretest probability of IPF (over 60 years of age, no signs of connective tissue disease, no exposure history, or pertinent medication history), a presumptive diagnosis of IPF can be made with a probable UIP pattern on CT, decreasing the importance of CT honeycombing from a diagnostic standpoint. Several studies have also shown that patients without honeycombing (probable UIP or indeterminate for UIP pattern) have a longer survival than those with honeycombing (UIP pattern).
HRCT Findings Suggestive of Alternative Diagnosis
HRCT findings that are suggestive of an alternative diagnosis include upper or mid-lung predominance, peribronchovascular or perilymphatic predominance, predominant ground-glass abnormality, profuse nodules, discrete cysts, marked mosaic attenuation/air trapping, and consolidation. Other findings suggestive of an alternative diagnosis include pleural plaques (indicating possible asbestosis), dilated esophagus or distal clavicular erosions (indicating connective tissue disease), extensive lymph node enlargement, pleural effusions, and pleural thickening. If any of these features are present in a patient with suspected IPF, the HRCT findings should be classified as “alternative diagnosis,” and surgical lung biopsy should again be considered. This does not necessarily mean that the diagnosis is not UIP, however, as there are a substantial number of cases that have an alternative diagnosis pattern on HRCT but are shown to have a UIP pattern on surgical lung biopsy and are eventually diagnosed as IPF.
Atypical Appearance on HRCT
An atypical HRCT appearance of UIP, either an indeterminate for UIP or an alternative diagnosis pattern on HRCT but a UIP pattern on histopathology, is fairly common. In one study of 55 biopsy-proven UIP cases, 62% of these cases were considered to have a low probability of representing UIP. Of these atypical UIP cases, the most common first-choice diagnoses based on HRCT were nonspecific interstitial pneumonia (NSIP), chronic HP, and sarcoidosis. Often, HRCT demonstrates peripheral basal-predominant reticular and ground-glass opacities with traction bronchiectasis but without significant honeycombing, an appearance commonly associated with fibrotic NSIP ( Fig. 19.5 ).


HRCT Features and Diagnosis of Pulmonary Fibrosis
HRCT features of pulmonary fibrosis include irregular pulmonary parenchymal interfaces, traction bronchiectasis and bronchiolectasis, regional volume loss, and honeycombing. Honeycombing is the most specific sign of fibrotic lung disease and results from alveolar disruption and dilation of bronchioles and alveolar ducts, with the creation of clustered, cystic air spaces lined by bronchiolar epithelium.
In the absence of honeycombing, pulmonary fibrosis can still be diagnosed by the presence of the other findings. There are often irregular interfaces at the edges of pulmonary vessels or bronchi, along the interlobar fissures, and along the peripheral pleural surfaces of the lungs. Traction bronchiectasis represents bronchial dilation in areas of pulmonary fibrosis secondary to the traction effect of the fibrous tissue on the bronchial walls. The bronchi often demonstrate an irregular or varicose morphology ( Fig. 19.6 ). In the extreme lung periphery of these patients, a dilated airway likely reflects traction bronchiolectasis. Over time, pulmonary fibrosis causes progressive volume loss, which is manifested by crowding of bronchovascular structures in areas of disease involvement and retraction of the fissures.


Differentiating Honeycombing From Traction Bronchiolectasis and Paraseptal Emphysema
On HRCT, honeycombing appears as cystic air spaces, several millimeters to several centimeters in diameter, with well-defined walls and predominating in a subpleural location ( Fig. 19.7 ). Honeycomb cysts typically share walls and occur in multiple layers, although early honeycombing may manifest as a single layer of subpleural cysts.