Interstitial pneumonia with autoimmune features (IPAF) refers to the clinical entity of interstitial lung disease (ILD) in patients with features of autoimmunity but without overt connective tissue disease (CTD). IPAF was introduced in 2015 by an international consensus panel in an attempt to standardize the nomenclature and diagnostic criteria for this entity, which represents an intermediary on the spectrum between the idiopathic interstitial pneumonias (IIPs) and CTD-associated ILD. The high-resolution computed tomography (HRCT) pattern is a critical component of IPAF criteria, and the prognostic significance of individual patterns and pattern features were recently described. As such, IPAF remains an area of active multidisciplinary investigation, and knowledge of this clinical entity is essential for a comprehensive understanding of ILD.
Background
ILD often arises in the context of an established CTD, such as rheumatoid arthritis, systemic lupus erythematosus, polymyositis, dermatomyositis, Sjögren syndrome, scleroderma, and mixed connective tissue disease. Nonspecific interstitial pneumonia (NSIP) is the most commonly encountered ILD pattern on HRCT in those with CTD, with the exception of rheumatoid arthritis, where a usual interstitial pneumonia (UIP) pattern is more commonly observed. Current international guidelines for the diagnosis of the IIPs recommend excluding CTD, which is typically performed by assessing for extrathoracic manifestations, autoantibody testing, and multidisciplinary discussion.
It is not uncommon, however, for ILD to be the first, or only, manifestation of autoimmunity. Many patients with ILD exhibit features of CTD but do not satisfy rheumatologic criteria for a specific CTD diagnosis. These patients have been previously described in the pulmonary medicine and radiology literature by various names, including: lung-dominant CTD, undifferentiated CTD-associated ILD, and autoimmune-featured ILD. The diagnostic criteria used to define these entities were overlapping but varied. Despite the associated features of autoimmunity, few of these patients will go on to develop overt CTD.
The need for a standardized nomenclature and diagnostic criteria for this patient population led to the formation of the European Respiratory Society (ERS)/American Thoracic Society (ATS) Task Force on Undifferentiated Forms of Connective Tissue Disease–Associated Interstitial Lung Disease. In 2015 this group produced an official ERS/ATS research statement proposing criteria for patients with IPAF. Patients with IPAF can be thought of as intermediary on the spectrum between the IIPs and those with overt CTD-associated ILD.
Diagnostic Criteria
The diagnosis of IPAF requires : the presence of ILD by HRCT or surgical lung biopsy, exclusion of an alternate etiology for the ILD, failure to meet criteria for a defined CTD, and at least one feature from at least two of these domains: clinical, serologic, and morphologic ( Table 45.1 ).
| CLINICAL DOMAIN | “Mechanic’s hands,” distal digital tip ulceration, inflammatory arthritis, polyarticular morning joint stiffness, palmar telangiectasia, Raynaud phenomenon, unexplained digital edema, Gottron sign | |
| SEROLOGIC DOMAIN | ANA ≥ 1 : 320, RF ≥ 2× upper limit of normal, anti-CCP, anti-dsDNA, anti-Ro (SS-A), anti-La (SS-B), anti-ribonucleoprotein, anti-Smith, antitopoisomerase (Scl-70), anti-tRNA synthetase, anti–PM-Scl, anti-MDA5 | |
| MORPHOLOGIC DOMAIN | HRCT | NSIP, OP, NSIP with OP overlap, LIP |
| Histopathology | NSIP, OP, NSIP with OP overlap, LIP, interstitial lymphoid aggregates with germinal centers, diffuse lymphoplasmacytic infiltration | |
| Multicompartment Involvement | Unexplained pleural effusion/thickening, unexplained pericardial effusion/thickening, unexplained intrinsic airways disease, unexplained pulmonary vasculopathy |
The clinical domain comprises features that are strongly associated with CTD. These include distal digital fissuring (“mechanic’s hands”), digital tip ulceration, inflammatory arthritis or polyarticular morning stiffness lasting more than 60 minutes, palmar telangiectasia, Raynaud phenomenon, unexplained digital edema, and unexplained fixed rash on the digital extensor surfaces (Gottron sign). The serologic domain includes autoantibodies with strong CTD association and requires moderately elevated titers for less specific autoantibodies, such as the antinuclear antibody (ANA) and rheumatoid factor (RF).
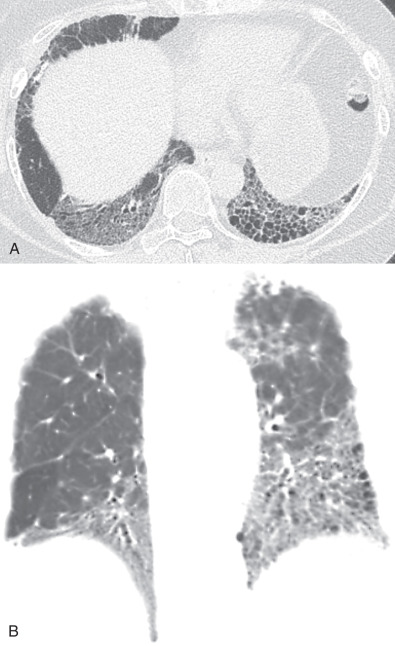
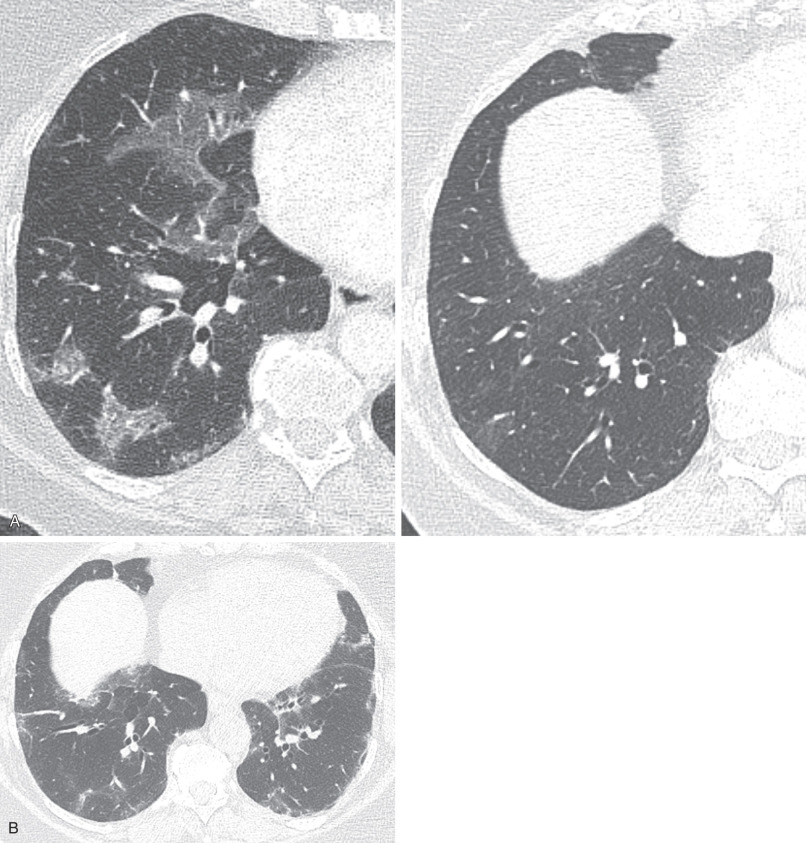
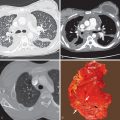
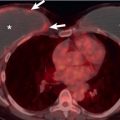
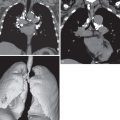
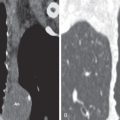
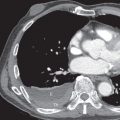
The morphologic domain includes three subdomains: radiologic, pathologic, and multicompartment. The radiologic subdomain is focused on HRCT features that are commonly associated with CTD, including NSIP ( Fig. 45.1 ), organizing pneumonia (OP) ( Fig. 45.2 ), NSIP with OP overlap ( Fig. 45.3 ), and lymphocytic interstitial pneumonia (LIP). UIP is not included in the IPAF criteria because it lacks specificity. The pathologic subdomain similarly includes NSIP, OP, NSIP with OP overlap, and LIP. Interstitial lymphoid aggregates with germinal centers and diffuse lymphoplasmacytic infiltration are also included because they are strongly associated with CTD. UIP is again not included because it lacks specificity for CTD.