Fig. 14.1
Diagram of normal eye anatomy in the axial plane. The anterior chamber, located anterior to the lens, extends from cornea to iris. The posterior chamber extends from the iris to posterior lens. Both chambers contain aqueous humor. Suspensory ligaments arising from the vascular ciliary bodies support the lens. The vitreous chamber extends from posterior lens surface to the ocular surface. The posterior ocular surface is comprised of three layers: inner retina, vascular choroid, and outer sclera. The ora serrata is the anterior most portion of the retina and covers the posterior two thirds of the inner surface of posterior chamber. The optic nerve extends posteriorly from the globe through retro-orbital fat. The central retinal artery and vein course through the optic nerve (adapted from [6])
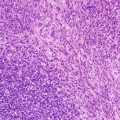
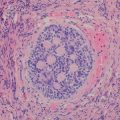
Fig. 14.2
Microphotograph of the retina with the name of the different layers. The pigmented epithelium has been cut tangentially and appears thicker. Bruch’s membrane separates the pigmented epithelium from the choroid
The eye structures derive from the ectoderm and mesoderm of the embryo. The optic pits appear as lateral evaginations of the neuroectoderm of the forebrain (prosencephalon) on day 22–23. As the forebrain closes, these evaginations continue to proliferate laterally and become large single layered vesicles (primary optic vesicles) that are continuous with the third ventricle. As the vesicles reach the surface ectoderm, they induce the formation of the lens placode. The optic vesicles invaginate, creating the optic cup with two layers. The differentiation of this secondary optic vesicle continues with the fusion of the retinal layers. The changes in the neural layers are similar to those in the brain. Rudimentary sensory cones and rods appear during the 12th week of gestation and continue development after birth. As the optic vesicles invaginate to form the optic cup and an inner and outer retinal layer, the point where the two layers meet anteriorly form the papillary opening. These two layers partially overlap the lens and become the iris at about 7th week of development. The ciliary body appears on the inner surface of the developing iris around week 9 and begins secreting aqueous humor [1].
Processing the Eye
Appropriate handling of the enucleated eye is fundamental in order to obtain appropriate fresh material for molecular genetic studies and to preserve the histology after fixation. In order to obtain material for genetic studies, the unfixed, fresh eye should be opened as soon as possible in order to minimize degradation of nucleic acids and proteins. The optic nerve is measured, and the resection margin is separated and fixed in a different cassette. The eye is oriented by localizing the inferior oblique muscle, which inserts in the sclera under the macular area and temporal to the optic nerve. Two basic techniques for the examination of enucleated eyes have been proposed by the Consensus meeting from The International Staging Work Group on the Pathology Guidelines [2]. First, the tumor is located with transillumination. In one approach, a window is opened in the sclera at the edge of the tumor. This can be done using a trephine or a sharp blade. Fresh tumor is retrieved from non-necrotic, viable areas, either with a scalpel and forceps or with a sterile pipette. The second technique proposed is the aspiration of tumor with a 22 G sterile needle under visual control through the sclera, using an approach posterior to the lens in a slightly oblique anteroposterior course. We use a modification of the first technique that allows for proper visualization of the tumor and preserves nucleic acids and proteins: after separating the optic nerve resection margin, we freeze the eye in liquid nitrogen for 20–25 s and then slice the superior calotte from the rest of the orbit. After the tumor thaws (after a few seconds depending on the size of the tumor), we extract fresh material with the help of a sterile scalpel or a sterile pipette, The tumor preservation for genetic analysis is perfect, and the technique introduces fewer artifacts in the histology (unpublished method). After harvesting fresh tumor, the eye is placed in sufficient formalin and fixed for 24–48 h.
After fixation the whole specimen is submitted for examination in four blocks: (1) optic nerve resection margin, (2) superior calotte, (3) mid section of the eye with the optic nerve and anterior chamber structures, and (4) inferior calotte. The calottes may be further sectioned into segments embedded on edge to examine more choroidal surfaces. Several levels may be necessary, particularly with small retinoblastomas after chemotherapy and for optimal assessment of optic nerve head invasion.
Overview of Diagnostic Imaging Approaches
Ultrasound (US) and magnetic resonance imaging (MRI) are the preferred imaging modalities to assess ocular lesions in children. Computed tomography (CT) is discouraged for several reasons: damage caused by the inherent exposure to ionizing radiation and the association of retinoblastoma with radiation-induced secondary malignancies. The latter factor is a particular consideration in children who are ultimately diagnosed with hereditary retinoblastoma. When assessing ocular disease, US can identify calcification within lesions, delineate tissue interfaces, and reveal patterns of vascularity using Doppler technology. Magnetic resonance imaging is less sensitive for the detection of calcification but more sensitive to tumor extension into the optic nerve and subarachnoid spaces. Therefore, these modalities are complementary and, in current practice, the combination of US, MRI, and fundoscopy obviates the need for CT in the setting of pediatric ocular tumors [3, 4].
An awareness of the imaging appearances of normal eye anatomy is necessary to accurately evaluate ocular lesions with diagnostic imaging. The US and MRI anatomy of the normal eye have previously been well described [5–7]. Ultrasound can be performed with conscious patients, and a typical examination takes 10–15 min. If needed, it US can be performed while the patient is sedated for MRI. A linear, high-frequency transducer with a small footprint is placed directly on ultrasound gel applied to the closed eyelid. The US machine’s mechanical index should be kept ≤0.23 to conform to FDA guidelines and to minimize risk of thermal injury to the eye [8]. Color Doppler is used to assess tumor vascularity, to determine the relationship of the mass to the optic disk, and to distinguish dense subretinal fluid from a solid mass. MRI requires some level of sedation for optimal imaging. Guidelines for standardized MRI of the eye were recently published [9]. In general MR examinations should include dedicated orbit imaging and imaging of the entire brain. Orbital imaging should include gadolinium-enhanced fat-suppressed sequences to optimize the delineation conspicuity of the tumor within orbital fat. The use of high-field strength magnets and surface coils may improve definition of the lesion and detection of optic nerve involvement. By using conventional coils, current 3-Tesla MR scanners provide improved imaging with a wider field of view as compared to surface coils [3, 10].
Retinoblastoma
Retinoblastoma, a rare tumor of the eye, characteristically arises in the retina of children and represents the most common orbital malignancy in childhood. Retinoblastomas occur primarily in children under the age of 5 years; the median age of diagnosis is 2 years of age [11] for unilateral retinoblastoma and 9–12 months for bilateral retinoblastoma. The disease is unilateral in 60 % of cases and bilateral in 40 %. All bilateral and 15 % of unilateral retinoblastomas are hereditary.
The incidence of retinoblastoma is 1 in 15,000–20,000 live births. The world incidence and mortality differs among countries (Table 14.1). A higher incidence of unilateral RB is seen in less developed countries, suggesting that environmental factors associated with poverty increases the risk of retinal mutations [12].
Table 14.1
World incidence and mortality for RB (Data from Shields and Shields 2012)
Location | Incidence | Patients/year | Mortality rate | Deaths/year |
|---|---|---|---|---|
Africa | 34.2 | 1,792 | 70 | 1,254 |
Asiaa | 18.8 | 4,027 | 39 | 1,591 |
Japan | 8.3 | 59 | 3 | 2 |
Europe | 10.2 | 414 | 5 | 21 |
North America | 13.8 | 258 | 3 | 8 |
Latin America /Caribbean | 19.8 | 622 | 20 | 124 |
Oceania | 16.3 | 21 | 10 | 4 |
Total | 7,202 | 3,001 |
Hereditary retinoblastoma is characterized by the presence of a germ line mutation, earlier clinical manifestation, and an elevated risk of developing secondary malignancies including osteosarcoma, malignant melanoma and soft tissue sarcoma [13]. Bilateral retinoblastoma arises when a germ line mutation in one copy of the RB1 gene is followed by a somatic mutation of the remaining wild copy. In non-hereditable RB both mutations are somatic.
Genetics
Retinoblastoma was the first malignancy to be described as a genetic disease. In 1971 Alfred Knudson proposed that retinoblastoma was initiated by inactivation of a tumor suppressor gene and that two events were necessary for the development of the disease [14]. This hypothesis was subsequently confirmed by the demonstration of the loss of heterozygosity at 13q14 in retinoblastomas [15] and the cloning of the first tumor suppressor gene RB1 [16]. Since then many tumors have been found to have defects in their RB pathway, through genetic lesions in either the RB1 gene or other genes in the pathway [17]. Retinoblastomas are initiated by the loss of both alleles of RB1 (M1 and M2, first and second mutational events). This is necessary but not sufficient for the development of the malignancy. The M1 event can be inherited or arises sporadically in a susceptible retinal cell, while M2 is always sporadic. A number of subsequent mutational events eventually lead to retinoblastoma [18].
In humans, the RB1 gene is located within a 183-kB region of chromosome 13q14 and is made up of 27 exons, which produce a 4.7-kB mRNA that is translated into a 928 amino acids protein called pRB. RB1 gene is a tumor suppressor gene and as such its protein product can inhibit tumor formation in a variety of tissues. pRB is a negative regulator of the cell cycle and has a role in cell differentiation, cell aging, apoptosis, and growth suppression. It is modified by phosphorylation, which affects the physical interaction between pRB and the E2F proteins that activate genes involved in the G1-S phase of the cell cycle and promote cell cycle progression. Hypophosphorylated pRB can bind to E2Fs, inhibiting this function. As pRB is phosphorylated, it loses its ability to bind to E2Fs, allowing them to activate the genes that promote the cell cycle. RB also has a role in suppression of apoptosis and indirectly controls differentiation [19].
Most retinoblastomas display genomic imbalances, the most frequent being gains of chromosomes 1q, 2p, and 6p and loss of chromosome 16q. Comparative genomic hybridization (CGH) reveals that over 50 % of retinoblastomas show a gain of 1q. The gains involve KIF14 (1q), a mitotic kinesin [18]; E2F3 and DEK (6p) the former a regulator of the cell cycle and DEK and the latter a known oncogene in many cancers [20]; and MYCN (2p) an oncogene amplified in a subset of neuroblastomas [17]. Approximately 30 % of retinoblastomas show a loss at chromosome 16q22 a region that contains two potential tumor suppressor genes belonging to the cadherin family [21]. Using a microarray analysis, 1,404 genes were upregulated and 481 genes were downregulated in retinoblastomas [22].
Loss of RB1 in many tissues leads to apoptosis; however, in the retina RB1-negative retinoblasts can survive and form tumors. A possible pro-apoptotic tumor suppressor gene in the retina is NGFR, which encodes p75, a modulator of cell death in the retina [23].
A small percentage of retinoblastomas are caused by deletions in the region of chromosome 13 that contains the RB1 gene. Because several other genes are also affected, these children usually have intellectual disability, slow growth, and distinctive facial features (prominent eyebrows, short nose with broad nasal bridge, and ear abnormalities) [24].
Genetic counseling is very important for retinoblastoma patients and should be offered to every parent with a child with retinoblastoma and to patients with a familial history of retinoblastoma. The nature of the disease, the inheritance patterns, and the risk to family members should be explained.
Both RB1 alleles have to be deregulated in order for a retinoblastoma to develop. However, the mutant allele behaves as an autosomal dominant trait. The majority of retinoblastomas (85–90 %) arise sporadically in the retina of the affected patient or in the parental germ line; there is no family history of the disease. If the initial RB1 mutation arises in the germ line before conception or in the early embryo, the risk for retinoblastoma will be heritable. If two RB1 somatic mutations occur in a retinal cell but the germ line is not affected, the tumor will not be heritable. In 88 % of sporadic bilateral retinoblastomas, there is a new mutation arising in the germ line of the father (>90 %) or the mother (<10 %). The remaining 12 % have mutational mosaicism, and the tumor may be heritable if the mutation is present in the germ line [25]. Sporadic tumors are more likely to be unilateral and unifocal; however, bilateral and multifocal tumors have also been observed. In addition, RB1 mutation predisposes to other primary cancers as detailed above. These secondary malignancies present later in life but earlier than in persons with no RB1 mutations. In the absence of molecular diagnosis, it is important to screen all first degree relatives of retinoblastoma patients when the first case is identified in the family.
In hereditary retinoblastoma, genetic testing of constitutional DNA (usually from a peripheral blood sample) identifies a large percentage of mutations in hereditary cases [26]. The second mutation can only be identified in tumor DNA. For sporadic bilateral retinoblastoma, analysis of peripheral blood may not always lead to the detection of the RB1 mutation. In these cases, testing of tumor DNA may show two RB1 mutations or one mutation and loss of heterozygosity. If a causative mutation is detected as heterozygous in the proband’s blood, the parents should be tested for the mutation. Most cases of sporadic retinoblastomas arise de novo, but in a small number the parent may be a low level mosaic. RB1 mutations can be prenatally detected in DNA from the amniotic fluid. For sporadic unilateral retinoblastoma, the peripheral DNA is usually normal (85 % of patients), and DNA from the tumor must be analyzed to identify both RB1 mutations.
Over 1,000 mutations leading to retinoblastoma have been identified, and a variety of techniques are used to find the nature of an unknown RB1 mutation [23].
Clinical Presentation
The most common presentation of retinoblastoma is leukocoria (white pupil), generally detected by family members, frequently on a flash photograph. This may present with or without strabismus [27]. Leukocoria may be recognized at a pediatric visit. Retinoblastoma generally remains intraocular and curable for 3–6 months after the first sign of leukocoria [23].
Less common forms of presentation include red eye, amblyopia, anisocoria, heterochromia, and hyphema. Patients with advanced disease may present with orbital cellulitis, particularly when there is significant tumor necrosis within the eye. Proptosis is a common presentation in low income countries but is rare in developed countries [28].
Patients with a known familiar predisposition are diagnosed earlier, as these children are screened after birth. Patients at high risk can be screened in utero [29].
There are many conditions that can mimic retinoblastoma. The three most common conditions that can simulate retinoblastoma are Coats’ disease, an abnormality of the retinal vasculature of unknown origin; persistent hyperplastic primary vitreous; and toxocariasis (Table 14.2).
Table 14.2
Differential diagnosis of retinoblastoma
Coat’s disease |
Persistent hyperplastic primary vitreous |
Toxocariasis |
Cellulitis |
Metastasis |
Coloboma |
Toxoplasmosis |
Retinopathy of prematurity |
Cytomegalovirus and herpes retinitis |
Astrocytic hamartoma |
As highlighted above, there is a high risk of secondary malignant tumors in patients with hereditary retinoblastoma who are born with a germ line mutation of the RB1 gene. Although more frequent after radiotherapy treatment, they may occur without radiotherapy and in children treated with chemotherapy [29, 30]. Chemotherapy is also known to enhance the development of second malignant tumors in irradiated patients [31]. Secondary tumors reported include osteosarcoma (the most common secondary malignancy both in irradiated and non-irradiated areas); soft tissue sarcomas (including leiomyosarcoma, fibrosarcoma, malignant fibrous histiocytoma, rhabdomyosarcoma, unspecified sarcomas, and liposarcoma, and closely associated with the use of radiation); skin cancer (melanoma and basal cell and squamous cell carcinoma, especially in the radiation area); common adulthood cancers (lung, breast, colon, oral cavity, bladder, uterus), hematological malignancies [13, 31–34], and sinonasal PNET [35].
The association of bilateral retinoblastoma with a synchronic or metachronic midline intracranial tumor is referred to as trilateral retinoblastoma (TRB), a rare but well recognized syndrome. Tumors of the suprasellar region tend to present earlier than tumors of the pineal region after the diagnosis of intraocular tumors. The prognosis is very poor. As there is better overall survival in children who are asymptomatic at the time of diagnosis of the intracranial tumors, screening is essential in patients with bilateral and/or hereditary retinoblastoma [36]. Since the use of systemic chemotherapy there has been a decrease in the number or TRBs. Pineal and parasellar tumors may appear several years after successful treatment of intraocular retinoblastoma and they may be better differentiated. The difference between TRB and metastatic tumor is that the latter presents as multiple undifferentiated tumors within the first 2 years of the in initial treatment
Imaging Features
The imaging appearance of retinoblastoma somewhat depends on whether the tumor is endophytic, exophytic, or diffusely infiltrating. Endophytic tumors arise from the inner layers of the retina and grow into the vitreous body. Small clusters of tumor may separate from an endophytic tumor to produce vitreous seeding. Exophytic tumors arise in the outer retinal layers and grow in the subretinal space causing retinal detachment, subretinal exudates, and possible subretinal tumor seeding. However, retinal detachment and subretinal fluid can occur in both endophytic and exophytic tumors. Therefore, these findings are of limited value in predicting the tumor growth pattern [9]. Diffusely infiltrating retinoblastoma, characterized by diffuse infiltration of the retina without a focal mass, is rare [4]. In contrast to the other types of retinoblastoma, the infiltrating type usually lacks calcification [3].
Ultrasound
Sonographically retinoblastoma appears as an irregularly shaped soft-tissue mass, more echogenic than the vitreous, with numerous hyperechoic foci due to focal calcification or microcalcifications (Fig. 14.3a). The presence of calcification is the hallmark of retinoblastoma and the key feature in distinguishing it from other ocular pathologies [4, 5]. With modern high-resolution US transducers, calcification is identified in 92–95 % of cases when it is found histologically. This compares to a sensitivity of 81–96 % for CT [4]. Retinal detachment is frequently present and appears as a thin strand of juxtatumoral material that is contiguous with the retina (Fig. 14.3a). Fluid in the subretinal space or vitreous is often slightly echogenic due to blood products, increased globulin content, or tumor seeding [6]. Cystic areas within the tumor are likely due to necrosis, a common pathologic finding. Ultrasonography is not useful for determining invasion of the optic nerve, which is often obscured by overlying tumor or tumor calcification (Fig. 14.3a) [4].
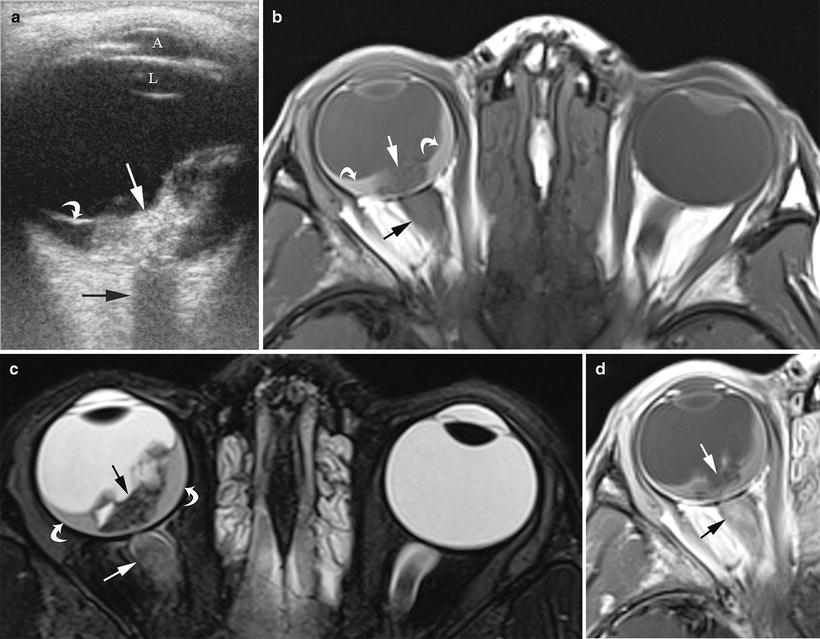
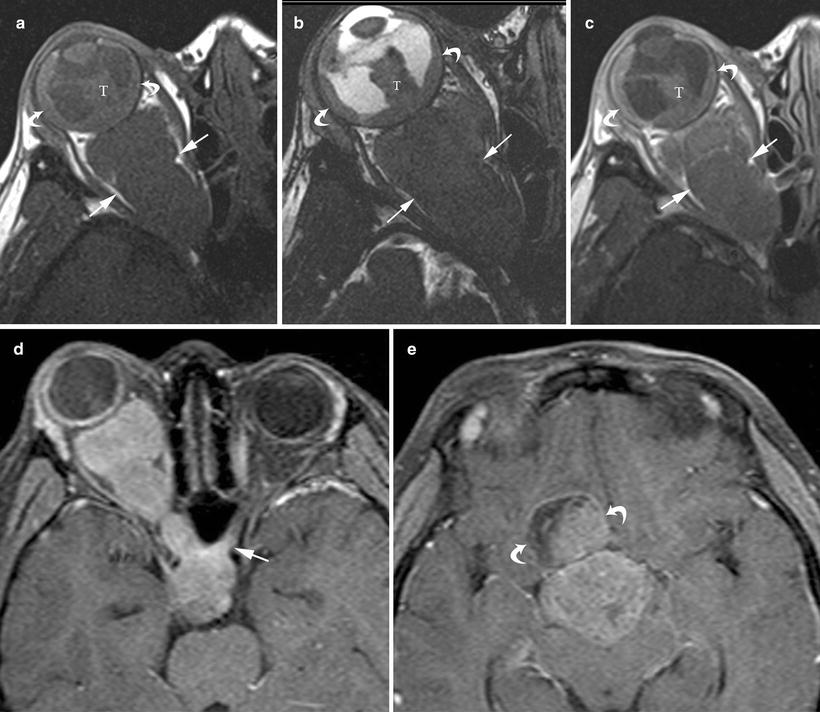
Fig. 14.3
A 5-year-old boy presented with right eye blindness and pain, found to have retinoblastoma. (a) Transverse ultrasound image shows dense calcification of the tumor (bright flecks, straight white arrow) causing posterior shadowing (black arrow) that obscures visualization of the optic nerve. Detached retina (curved white arrow) is evident. A anterior chamber, L lens. (b) Axial, non-contrast enhanced T1W, (c) axial T2W and (d) contrast-enhanced T1W MR images show the primary tumor to be of mixed, but predominantly low signal intensity on non-contrast T1W imaging (straight white arrow) and T2W (straight black arrow), with minimal enhancement probably due to dense calcification. Subretinal fluid (curved arrows) is hyperintense to vitreous on T1W and hypointense on T2W images. The optic nerve (straight black arrow on images (b) and (d), straight white arrow, image (c) is thickened relative to the normal left side and enhances inhomogenously. On pathologic inspection this tumor was densely calcified, mixed endo-exophytic with minimal choroidal involvement, detached retina, subretinal hemorrhage and gliosis, and extensive optic nerve invasion
Magnetic Resonance Imaging
On MRI, retinoblastoma typically matches the signal intensity of gray matter. On T1W images the tumor is slightly hyperintense to vitreous, and on T2W images it is usually darker than vitreous. The tumor may appear heterogeneous due to internal calcification. Retinoblastomas typically demonstrate moderate, heterogeneous contrast enhancement (Figs. 14.3b–d, 14.4a–c). Magnetic resonance imaging can detect ocular lesions as small as 3 mm in size, but as vitreous seeds rarely exceed 1–2 mm they may be missed by MRI [3, 9, 10]. MRI has a sensitivity of 50–89 % for detection of retinal detachment; it can also detect sub-retinal fluid and vitreous hemorrhage. Subretinal and vitreous fluid may contain fluid–fluid levels due to sedimentation effect and may be hyper- or isodense. They may appear uniformly hyperintense on both T1 and T2W images due to hemorrhage or proteinaceous subretinal effusion, or they may appear isointense to vitreous on T1W images and hyperintense on T2W images due to subretinal fluid with low protein content (Fig. 14.4b–d) [9]. Retinoblastoma can breach the Bruch membrane and infiltrate the choroid (Figs. 14.4a–c, 14.5b–d) [3]. Using standard head coils, homogenous enhancement and thickening of the choroid adjacent to the tumor is 73 % sensitive and 72 % specific for choroidal involvement by MRI. Choroidal inflammation can mimic the appearance of tumor involvement on MRI, thus limiting its utility in this regard. Once the choroid has been infiltrated, tumor can spread along ciliary vessels and nerves to extend into the sclera, orbit, and conjunctiva, findings which may be apparent on MRI (Fig. 14.4a–c) [3]. Magnetic resonance imaging has been shown to have a sensitivity of 66 %, specificity of 96 %, and accuracy of 79 % for prelaminar optic nerve involvement (anterior to the insertion of the meninges on the optic nerve). Regarding post-laminar optic nerve involvement, MRI is reported to have a sensitivity of 50 %, specificity of 100 %, and accuracy of 97 % [9]. On post-contrast images, prelaminar optic nerve involvement appears as interruption or thickening of linear enhancement at the choroidal retinal complex. Enhancement extending from the tumor into the optic nerve is a reliable sign of postlaminar optic nerve involvement, but its absence does not preclude optic nerve involvement (Figs. 14.3b, c, 14.4c, 14.5a–d). Both pre- and post-laminar microscopic invasion of the optic nerve can be missed by MRI, so that primary tumor volume is thought to be the best predictor of optic nerve invasion [9]. Once extraocular spread occurs, the risk of distant hematogenous spread to the lungs, bones, brain, and local-region lymph nodes increases [3]. Because of the risk of intracranial tumor extension or pineal tumors (“trilateral” retinoblastoma, which occurs with bilateral eye tumors), MRI of the orbits and brain should be obtained in the same scanning session. If intracranial extension is present, MRI of the entire spine is indicated in order to detect leptomeningeal involvement [3].
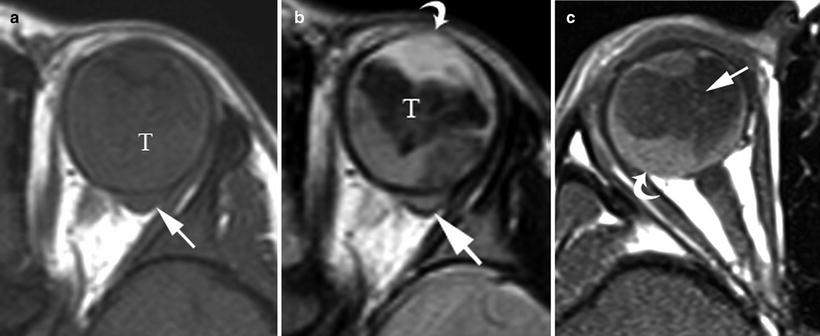
Fig. 14.4
A 3-year-old boy presented with leukocoria of left eye and was subsequently diagnosed with retinoblastoma. (a) Non-contrast enhanced axial T1W, (b) axial T2W and (c) post-contrast enhanced axial T1W MR images show the primary tumor expanding the left globe (note enlargement of left globe relative to normal right globe in (c)). Tumor (T) invades the anterior chamber (curved arrow (b)) and invades through the posterior sclera (straight white arrow (a, b) and straight arrow (c)) and the optic nerve (straight white arrow (c)). There is enhancing tumor causing retinal detachment (curved arrow (c)) and largely non-enhancing tumor (T) that is hypointense on T1W and T2W images probably due to calcification or hemorrhage. At pathologic inspection the tumor was mixed endo-exophytic with massive choroidal involvement, involvement of the vitreous, ciliary bodies, iris, anterior chamber, sclera, cornea and optic nerve with retinal detachment