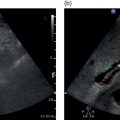
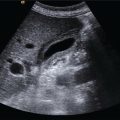
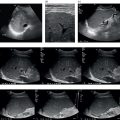
Andreas Panayiotou1, Maria E. Sellars1, and Annamaria Deganello1,2 1 Department of Radiology, King’s College Hospital, London, UK 2 Division of Imaging Sciences, King’s College London, London, UK Paediatric liver disease is different from liver disease in the adult population, in terms of aetiology (more often genetic or congenital in children versus secondary to environmental exposure in adults) and susceptibility and response to injury; therefore numerous pathologies are unique to this age group. In this chapter, the emphasis will be on hepatobiliary scanning technique in the neonatal and paediatric age group, as well as the ultrasound appearance of the most common diffuse and focal liver pathologies. Ultrasound is the first‐line method of imaging of the liver in the paediatric population for obvious reasons: it is relatively inexpensive, does not require sedation, does not involve ionising radiation, and can easily be performed at the patient’s bedside (i.e. in the paediatric and neonatal intensive care unit), next to the parents. To perform ultrasound on children, a high‐frequency transducer is used for high‐resolution anatomical detail. High‐frequency linear probes can be used in neonates to assess the gallbladder and biliary tree, and high‐frequency curvilinear probes should be used in older children and to obtain Doppler traces of the hepatic vasculature. A systematic approach is needed with a detailed assessment of the right upper quadrant, including the liver, gallbladder, bile ducts, and portal vein. The ultrasound examination should also include the spleen and the lower abdomen and pelvis, to assess the presence of any associated congenital findings or complications, such as ascites. The liver parenchyma and size need to be assessed as well as the texture of the liver parenchyma. The normal hepatic parenchyma, similarly to adults, is smooth and homogeneous, and the peripheral portal vein branches should be clearly seen, with well‐defined, echogenic walls, as opposed to the thin‐walled hepatic veins. The gallbladder shape and size should be examined as well as the presence of biliary duct dilatation; the pancreas and pancreatic duct should also be included in the investigation. Calculi, inspissated bile, pericholecystic fluid, and sonographic Murphy’s sign should also be assessed. Although normal anatomy is similar to adults, it should be remembered that measurements differ according to paediatric age. The common bile duct is <1 mm in infants to 1 year of age, <4 mm in older children, and <7 mm in adolescents and young adults [1]. The pancreatic duct measures 1–2 mm and the gallbladder length is 1.5–3 cm in infants up to 1 year of age, and 3–7 cm in older children. Normal splenic length ranges from <6 cm in infants up to 3 months of age and <12 cm in older children [2]. Biliary atresia is a congenital absence or deficiency of the extrahepatic biliary system which can lead to neonatal cholestasis. If left untreated, it will lead to subsequent biliary cirrhosis, end stage liver disease and death. Therefore early diagnosis is important to achieve a favourable outcome. There are various ultrasound findings that can aid in the diagnosis of biliary atresia. The most sensitive signs include the triangular cord sign (hyperechoic triangular or tubular fibrous tissue along the anterior wall of the portal vein at the porta hepatis) and an abnormal, often small gallbladder with irregular walls (Figure 10.1a). Other findings may include a hypertrophic, hyperdynamic hepatic artery, a cyst at the porta hepatis, asplenia, absent gallbladder, non‐visible/absent common bile duct, and sub‐capsular flow [3]. Biliary atresia can be associated with other congenital abnormalities such as biliary atresia splenic malformation (BASM) syndrome (Figure 10.1b), for which these signs should be sought: polysplenia, situs inversus, portal vein abnormalities (hypoplastic, pre‐duodenal portal vein), interrupted inferior vena cava (IVC) with an azygous continuation, and intestinal malrotation. Patients with biliary atresia should also have a formal cardiac assessment with an echocardiogram to look for cardiac defects [4]. Treatment of biliary atresia entails a surgical procedure to restore bile flow known as ‘Kasai portoenterostomy’. Regular ultrasound follow‐up of these patients are imperative in order to detect the appearance of any sign of progression to chronic liver disease as a consequence of failure of the procedure. Thus, ultrasound assessment of these patients will aim to highlight signs of portal hypertension such as portal venous flow velocity reduction, increased hepatic artery resistance index and splenomegaly. A thorough liver parenchymal evaluation is also warranted to exclude the presence of focal lesions. Of note, increased liver stiffness, estimated by elastography, has been reported as an early predictor of Kasai procedure related complications and failure (Chapter 9.2). Figure 10.1 A 9‐month‐old infant with biliary atresia and splenic malformation (BASM) syndrome, with situs inversus. B‐mode ultrasound demonstrates a hyperechoic fibrous ductal remnant of the extrahepatic bile duct, ‘triangular chord sign’ at the bifurcation of the portal vein (a, arrows), and polysplenia (b, arrows). It is important to highlight that years after the Kasai procedure, the liver architecture may undergo remodelling in the form of central plate hypertrophy (and hence heterogeneity). Also known in the past as arteriohepatic dysplasia, Alagille syndrome is a rare genetic multiorgan systemic disorder characterised by a paucity of bile ducts and chronic cholestasis. It is diagnosed by demonstrating a paucity of intrahepatic bile ducts on liver biopsy and other criteria such as abnormal facies, congenital heart defect, ocular abnormalities, and vertebral anomalies [5]. Alagille syndrome can present with jaundice and hyperbilirubinemia and shares similar findings with biliary atresia and neonatal hepatitis. It is important to differentiate early, because biliary atresia requires early surgical management while Alagille can be managed conservatively. However, 15% of children with Alagille would require liver transplantation due to progression to end‐stage liver failure and refractory symptoms. Alagille is principally a clinical diagnosis and ultrasound findings are non‐specific and similar to biliary atresia. Both biliary atresia and Alagille syndrome can have evidence of an abnormal gallbladder shape and hypertrophy of the hepatic artery, although in Alagille the triangular cord sign is not present and hypertrophy of the hepatic artery is less commonly seen; in addition, in Alagille signs of portal hypertension are less common [6]. Alagille syndrome can also be differentiated from biliary atresia by the presence of associated congenital abnormalities such as abnormal facies and other visceral anomalies. Ultimately, biopsy and genetic testing are required for diagnosis [5]. Also known as ductal plate malformations, fibropolycystic liver disease is a complex group of congenital liver diseases resulting from abnormal embryonal development of the bile ducts. These disorders include congenital hepatic fibrosis, biliary hamartomas, polycystic liver disease, choledochal cysts, and Caroli disease, and can be associated with autosomal recessive polycystic kidney disease (ARPKD), medullary sponge kidney, and nephronophthisis. They constitute a spectrum of disorders that can co‐exist in the same patient and their manifestation and radiological signs will depend on the timing by which they occur during embryogenesis and the size of the bile ducts affected, with hepatic fibrosis being most commonly seen in abnormalities of the small, peripheral bile ducts [7]. Congenital hepatic fibrosis is characterised by periportal fibrosis, which can be seen on ultrasound as increased periportal echogenicity, and often presents in adolescents with signs of portal hypertension, such as splenomegaly and varices. Other ultrasound findings include hypertrophy of the left liver lobe, especially segment IV, with right lobe atrophy, and a hypertrophic hepatic artery with possible development of large intrahepatic regenerative nodules. Renal abnormalities such as polycystic kidney disease and medullary sponge kidney as well as co‐existing Caroli disease are often seen in association with this. Biliary hamartomas, also known as ‘von Meyenburg complexes’, are small scattered collections of dilated intrahepatic bile ducts that have lost their communication with the biliary tree and are distributed evenly throughout the liver parenchyma, measuring up to 1.5 cm in size. On ultrasound, they can present as multiple small hypoechoic or hyperechoic lesions with comet‐tail artefacts (See Chapter 6). Polycystic liver disease is a group of genetic disorders that can present in isolation, as autosomal dominant polycystic liver disease (ADPLD), or in combination with autosomal dominant or recessive polycystic renal disease (ADPRD or ARPRD). The disease is characterised by cysts that progressively increase in number and replace the liver parenchyma. Patients with polycystic liver disease show on ultrasound examination an enlarged liver, with bi‐lobar thin‐walled cysts of various sizes, ranging from a few millimetres to several centimetres in diameter. Possible complications include infection, compression of biliary ducts (even though the cysts do not communicate with the biliary tree), and bleeding or rupture of the cysts. Caroli disease and Caroli syndrome are seen on imaging as multifocal segmental dilatation of the large intrahepatic bile ducts, which still preserve their communication with the biliary tree. In Caroli disease, the arrest of remodelling of the ductal plate affects the larger intrahepatic ducts, whereas in Caroli syndrome, the arrest occurs both early and late during embryogenesis; in the latter, there is almost invariably a concomitant degree of congenital hepatic fibrosis. On ultrasound there is saccular or fusiform dilatation of the intrahepatic bile ducts (Figure 10.2), which may contain inspissated bile or stones. Typical of the disease is the ‘central dot sign’, better appreciated on magnetic resonance imaging (MRI) but still visible on ultrasound, where a central portal venous radicle and hepatic artery are surrounded by the dilated bile duct. Caroli can be differentiated from primary sclerosing cholangitis by the absence of focal strictures. Figure 10.2 A 3‐year‐old with Caroli syndrome. (a) B‐mode image of the liver demonstrates multiple, anechoic saccular dilatations of the intrahepatic bile ducts, with a background of heterogenous liver parenchyma in keeping with congenital hepatic fibrosis. (b) B‐mode image of the left kidney demonstrates numerous anechoic cysts and hyperechoic parenchyma in keeping with polycystic kidney disease. Choledochal cysts consist in dilatation of the intra‐ and/or extrahepatic bile ducts, and different theories have been proposed to explain their aetiology, including one that sees them as part of ductal plate malformation. A more common theory is that they are caused by reflux of pancreatic enzymes and cholangitis due to an abnormal pancreaticobiliary junction with a long common channel, which eventually results in dilatation of the biliary system [8]. To make a diagnosis of a choledochal cyst, there must be a demonstrable communication with the biliary duct system. On ultrasound, choledochal cysts can be seen in different sizes and locations, according to the revised Todani classification (Table 10.1) [9, 10]. The pancreas should be assessed for duct dilatation and pancreatitis. Other complications include abscess, cirrhosis, malignancy, gallbladder, and biliary tract stones, as well as biliary peritonitis from spontaneous rupture of the cyst. Progressive familial intrahepatic cholestasis (PFIC) is a rare genetic disorder in which the liver fails to adequately secrete bile, resulting in cholestatic jaundice and pruritus. There are three types of PFIC; all patients are at risk of developing portal hypertension, ascites, liver failure, and, especially in PFIC type 2, cirrhosis and liver cancer. Ultrasound is the imaging modality of choice to screen these patients mostly for the early detection of hepatocellular carcinoma (HCC), as well as signs of portal hypertension, gallstones, and complications such as pancreatitis. Cystic fibrosis–associated liver disease has become a well‐recognised complication of cystic fibrosis. Prompt identification and treatment can lead to better outcomes. The aetiology is an abnormal cystic fibrosis transmembrane regulator (CTFR) protein in the biliary epithelium, resulting in the accumulation of viscous bile. This results in injury to the hepatocytes and biliary epithelium, leading to fibrosis. Hepatic complications in cystic fibrosis are normally seen in older children; such children are usually asymptomatic until they present late with established portal hypertension and liver cirrhosis. Ultrasound may detect focal biliary fibrosis, which manifests as increased periportal echoes of heterogenous liver parenchyma. Additional findings include hepatomegaly, periportal fat deposition, biliary abnormalities including altered gallbladder morphology, and features of liver cirrhosis and portal hypertension (Figure 10.3) [11]. Table 10.1 Todani classification of bile duct cysts. Wilson’s disease is an uncommon inherited autosomal recessive disorder of copper metabolism. Excess copper accumulation results in chronic liver disease, although in some cases it can cause acute or even fulminant liver failure. Ultrasound findings include features of hepatic steatosis, acute hepatitis, and cirrhosis. Compared to other causes of chronic liver disease, in Wilson’s disease, the caudate lobe is not hypertrophied. A perihepatic fat layer can be observed and the outline of the liver may appear smooth or irregular with parenchymal hypoechoic or hyperechoic nodularities as well as a combination of both (mixed pattern) [12]. Acquired diffuse liver disease includes hepatitis of various causes. The most common causes in this age group are non‐alcoholic fatty liver disease (NAFLD), viral and autoimmune hepatitis, and sclerosing cholangitis. Diffuse liver disease often has non‐specific ultrasound findings ranging from normal to end‐stage liver disease. Ultrasound features of end‐stage liver disease include nodular liver parenchyma and features of portal hypertension, which are hepatofugal flow in the portal vein, venous collaterals, splenomegaly, and ascites (See Chapter 8). In the acute setting, ultrasound is mainly used as a screening tool to exclude obstructive causes of jaundice in patients who present with cholestasis (See Chapters 6 and 12). NAFLD is characterised by excessive build‐up of fat in the liver secondary to dysmetabolism or increased caloric intake in the absence of alcohol. There is an increasing prevalence of NAFLD in the paediatric population and it is one of the most common causes of chronic liver disease in children [13]. Steatosis is a hallmark of NAFLD and it is easily diagnosed with ultrasound by comparing the echogenicity of the right liver lobe to the cortex of the right kidney. In normal conditions the liver parenchyma demonstrates a smooth homogeneous echotexture with similar echogenicity to the renal cortex. Instead, when steatosis is present, the liver’s echogenicity appears increased compared to the cortex of the right kidney [14], and it can be graded subjectively according to the severity of posterior attenuation (See Chapter 8
10
Liver Ultrasound in the Paediatric Population
Diffuse Liver Disease: Congenital
Biliary Atresia

Alagille Syndrome
Fibropolycystic Liver Disease

Progressive Familial Intrahepatic Cholestasis
Cystic Fibrosis–Associated Liver Disease
Type IA
Dilatation of the entire extrahepatic bile duct
Type IB
Focal, segmental dilatation of the extrahepatic bile duct
Type IC
Smooth, fusiform dilatation of the extrahepatic bile duct
Type II
Diverticuli of the extrahepatic bile duct
Type III
Dilatation of the intraduodenal common bile duct (choledochocele)
Type IVA
Multiple dilatations of the intrahepatic and extrahepatic bile ducts
Type IVB
Multiple dilatations of the extrahepatic bile ducts only
Type V
Dilatation of the intrahepatic bile ducts (Caroli disease)
Wilson’s Disease
Diffuse Liver Disease: Acquired
Non‐alcoholic Fatty Liver Disease/Metabolic Dysfunction‐Associated Steatotic Liver Disease
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


