Etiology
Lymphangioleiomyomatosis (LAM) is a systemic disease of unknown etiology, affecting almost exclusively women. LAM is characterized by idiosyncratic smooth muscle cell proliferation (LAM cells), which leads to lung cysts, systemic lymphatic abnormalities, and abdominal tumors. The main manifestation is a progressive destructive process of the lungs, which may result in respiratory failure.
LAM cells can be isolated from peripheral blood, indicating the ability to spread hematogenously to the lung. LAM cells have been detected in donor lungs of patients who had lung transplantation, suggesting migration of LAM cells to the lungs from other sites, such as the kidney, lymphatic system, or uterus. Indeed, LAM is also defined as a low-grade, destructive, metastasizing neoplasm.
LAM can occur without evidence of other disease (sporadic LAM [S-LAM]) or in association with tuberous sclerosis complex (TSC). TSC is a neurocutaneous disease characterized by hamartomatous changes in the brain, kidneys, skin, heart, lungs, and other organs. TSC is an autosomal-dominant inherited disorder resulting from a germline mutation in one of two tumor suppressor genes, TSC-1 (encoding hamartin on chromosome 9q34) and TSC-2 (encoding tuberin on chromosome 16p13.3). In S-LAM somatic mutations have been found in LAM cells in the lungs, lymph nodes, and angiomyolipomas (AMLs), predominantly for TSC-2. S-LAM has been considered a forme fruste of TSC by some investigators, causing speculation that S-LAM results from somatic mutations associated with perinatal or early life events, possibly modified by hormonal factors.
Hamartin or tuberin deficiency or dysfunction results in upregulated activity of mammalian target of rapamycin (mTOR), which leads to increased protein translation and ultimately inappropriate cellular proliferation, migration, and invasion. Indeed, both sirolimus and everolimus—two mTOR inhibitors—have both demonstrated treatment benefit in LAM. These drugs are now recognized as the standard therapy for patients with declining lung function, symptomatic chylous effusions, lymphangioleiomyomas, or large AMLs. However, patients with advanced disease may require lung transplantation. The median transplant-free survival is approximately 29 years from the onset of symptoms and a 10-year transplant-free survival of 86%.
Disorganized lymphangiogenesis has an important role in the pathogenesis of LAM. An important biomarker of value in diagnosing LAM is vascular endothelial growth factor (VEGF)-D, a lymphangiogenic growth factor that is increased in the serum of patients with LAM, especially those with lymphatic involvement. In the appropriate clinical and radiologic setting, a VEGF-D serum level greater than 800 pg/mL is rarely found in other cystic lung diseases. VEGF-D may be of value in grading the severity of disease and monitoring therapeutic response.
Estrogen has also been implicated in the pathogenesis of LAM on the basis of the observation that LAM may be exacerbated by the administration of exogenous estrogens and during pregnancy or menstruation. Hormonal receptors are inconsistently expressed in affected tissues, however, and hormonal treatment has proved, at best, to be of only modest benefit.
Prevalence and Epidemiology
Prevalence of S-LAM is approximately 3.4 to 7.8 per million women in the United States and Europe. Most individuals who seek medical attention for LAM have S-LAM, as reported by the National Heart, Lung, and Blood Institute LAM Registry, which recorded the presence of TSC in 18.3% of 230 patients with pulmonary LAM. The expected prevalence of TSC-LAM should be higher than S-LAM because TSC affects 1 in 6000 children. This paradox of S-LAM being more common than TSC-LAM remains unresolved, but possible explanations include the suggestion that TSC-LAM may be less aggressive than S-LAM or that subclinical S-LAM is much more common than is currently appreciated. Mother-daughter transmission of TSC-LAM, but not S-LAM, has been documented, although two-thirds of cases of TSC-LAM are sporadic and likely are related to spontaneous mutations or incomplete penetrance.
Occurrence of cystic lung disease in women with TSC ranges from 26% to 49%. However, it was estimated that up to 80% of women older than 40 years with TSC will develop lung cysts. Lung cysts are not uncommon in men with TSC, affecting about 38%. Conversely, pulmonary involvement has been rarely reported in men with S-LAM.
LAM is usually diagnosed in women of childbearing age, with a mean age of 40 years. There are increasing reports, however, of women developing LAM after menopause, including women in their 80s.
- •
The etiology of lymphangioleiomyomatosis (LAM) is unknown.
- •
LAM occurs almost exclusively in women, usually during childbearing age.
- •
Prevalence of sporadic LAM is approximately 5 per million.
- •
Prevalence of TSC is 1 in 6000–9000.
Clinical Presentation
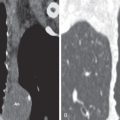
The most frequent presenting symptoms of patients with pulmonary LAM are dyspnea, often exacerbated by exertion, and fatigue. Dyspnea is due to airflow obstruction, replacement of the lung parenchyma by cysts, and, if the dyspnea is acute, pneumothorax. Pneumothorax is the sentinel event in 40% to 50% of patients ( Fig. 35.1 ). In one study of 395 patients, 260 patients reported at least one spontaneous pneumothorax (incidence, 66%), and 200 of 260 patients (77%) had subsequent pneumothoraces. It was shown that screening for LAM by computed tomography (CT) in women presenting with spontaneous pneumothorax can be cost-effective in an appropriately selected patient population (e.g., nonsmoking women, age 25–54 years, with a history of at least two episodes of pneumothorax and dyspnea before the pneumothorax).

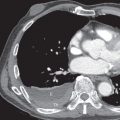
Other possible accompanying symptoms of pneumothorax are chest pain and cough. Pleural effusions occurring in patients with LAM are nearly always chylous. Chylothorax has been described in 14% of patients with LAM at presentation and in 22% to 39% during the course of the disease ( Fig. 35.2 ). Chylothorax may result from obstruction of the thoracic duct or its tributaries, leakage from pleural lymphatics, or transdiaphragmatic flow from associated chylous ascites. Less common manifestations are hemoptysis and chyloptysis (expectoration of milky chyle), which are likely due to LAM cell obstruction of pulmonary venules or capillaries and lymphatics. In rare cases patients with LAM may develop a chylous pericardial effusion (see Fig. 35.2 ). Cyanosis, respiratory failure, and cor pulmonale can occur in advanced disease. Physical examination is often normal at presentation, unless a pneumothorax or chylous effusion is present. Wheezing is heard in a few patients, and clubbing is a very rare accompaniment. Pulmonary hypertension secondary to chronic respiratory failure may be present in advanced cases of LAM. Abdominal examination may reveal the presence of ascites or masses, usually from AMLs or axial lymphatic involvement.

Occasionally, extrapulmonary manifestations are the first symptoms of LAM, most commonly bleeding from a renal AML. Multiple renal AMLs and AMLs greater than 4 cm are more likely to grow and cause symptoms, such as flank pain, hematuria, and, rarely, shock secondary to acute hemorrhage. Renal AMLs occur in approximately 93% of patients with TSC-LAM, and they are found in 32% to 60% of patients with S-LAM. In a study by Avila and colleagues, extrapulmonary manifestations of S-LAM were documented in 61 of 80 patients (76%) and included renal AMLs (43%), lymphadenopathy (39%), lymphangioleiomyomas (16%), ascites (10%), dilation of the thoracic duct (9%), and hepatic AMLs (4%). Lymphangioleiomyomas occur along the lymphatic system and may lie between, and displace, abdominal or pelvic structures ( Fig. 35.3 ). The most common symptoms associated with lymphangioleiomyomas are abdominal bloating, lower extremity edema, and urinary incontinence. These symptoms may worsen during the day because of an increase in size, likely owing to increased lower limb lymph accumulation as the day progresses. Overdistention of the fluid-filled lymphangioleiomyoma may result in rupture and chylous ascites. LAM is also associated with an increased frequency of meningioma.

Early reports of LAM suggested a pessimistic view of survival, with most patients dying within 10 years. More recent studies have reported a longer survival; however, this may reflect lead time bias as a consequence of earlier diagnosis. Some patients with LAM remain stable for years or decades, with only mild ventilatory impairment, whereas others experience rapid disease progression leading to respiratory failure or death within a few years of disease onset. The rate of decline in lung function tends to be greater in premenopausal women than in postmenopausal women. It has been suggested that greater age at presentation and presence of AML were associated with less risk of mortality. However, at present, there is still no good way of predicting the prognosis of an individual with LAM because the natural course varies.
Clinical features differ slightly among women with S-LAM and patients with TSC-LAM ( Table 35.1 ). Patients with TSC-LAM are more likely to be asymptomatic or to present with gradual onset of dyspnea, whereas patients with S-LAM are more likely to present with a pneumothorax. Chylothoraces occur less commonly in patients with TSC.
| S-LAM | TSC-LAM | |
|---|---|---|
| TSC-1 gene | − | + |
| TSC-2 gene | + | ++ |
| CLINICAL FEATURES | ||
| Dyspnea | ++ | + |
| Pneumothorax | ++ | + |
| Chylothorax | ++ | + |
| Asymptomatic | − | + |
| COMPUTED TOMOGRAPHY FEATURES | ||
| Pulmonary cysts | ++ | + |
| Nodules | − | + |
| Angiomyolipoma | + | ++ |
| Lymphangioleiomyomas | + | ++ |
| Lymphadenopathy | ++ | + |
TSC is a disorder with a high penetrance but variable expression. Not all patients have the complete triad of symptoms of TSC—epileptiform seizure, facial angiofibromas, and mental retardation—because half of all patients are of normal intellect, and a quarter do not have seizures. A panel of international experts considered the diagnostic criteria for TSC, recognizing that there are truly no pathognomonic clinical signs for TSC, and a detailed analysis of the hierarchy of clinical and imaging features necessary for a definitive, presumptive, or suspect diagnosis of TSC was undertaken. The latest diagnostic criteria consist of major features (e.g., cortical tuber, pulmonary LAM, facial angiofibroma, retinal hamartoma, renal AML) and minor features (e.g., gingival fibroma, hamartomatous rectal polyp, bone cyst). A diagnosis of TSC is definite when two major features or one major and two minor features exist, probable if one major and one minor feature are present, and possible when more than two minor features or only one major feature is present. When LAM and AML occur in the same patient, however, they should be considered as one major feature of TSC, not two.
- •
Dyspnea, fatigue, and pneumothorax are the most common initial manifestations of lymphangioleiomyomatosis (LAM).
- •
Patients with tuberous sclerosis complex–LAM tend to be less symptomatic than patients with sporadic LAM.
- •
Abdominal manifestations occasionally precede the diagnosis of pulmonary LAM.
Pathophysiology
Anatomy
On macroscopic examination, lungs affected by LAM are enlarged, with a striking appearance of subpleural blebs. The cut surface shows a honeycombing appearance owing to air-filled or fluid-filled (chylous or serosanguineous) cysts ranging in diameter from millimeters to centimeters.
Axillary, cervical, subclavian, mediastinal, retroperitoneal, and pelvic lymph nodes may be involved in LAM. The nodes are pale and spongy, tan or white. The thoracic duct may be enlarged, resembling a spongy sausage-like structure. Other gross findings include right ventricular hypertrophy, pleural adhesions, and chylous effusion.
Pathology
Two key microscopic features characterize LAM: cyst formation and proliferation of LAM cells ( Fig. 35.4 ). LAM cells express smooth muscle actin, desmin, and vimentin consistent with a smooth muscle lineage, although they also have features that are not typical of normal muscle cells—electron-dense granules, which contain melanoma-related proteins, including glycoprotein 100 (the target of the antibody HMB45) and tyrosinase and receptors for estrogen and progesterone (see Fig. 35.4 ). LAM lesions express VEGF-C and VEGF-D and often contain an abundance of lymphatic channels lined by vascular endothelial growth factor receptor (VEGFR)-3–expressing endothelial cells.

LAM cells tend to proliferate to form microscopic nodules, the centers of which contain predominantly spindle-shaped LAM cells, which show strong immunoreactivity for actin and focal positivity for melanoma-associated antigens, whereas the periphery comprises larger epithelioid LAM cells, which reveal the opposite phenotypic profile. LAM cells typically are found at the edges of the cysts and along pulmonary blood vessels, lymphatics, and bronchioles. Involvement of small pulmonary vessels is associated with thickened arterial walls and areas of venous occlusion, resulting in hemorrhage and hemosiderosis. In the lymphatics LAM cells form haphazard clumps, leading to thickening of lymphatic walls and, variably, obliteration of the vessel lumen and cystic dilation. Obstruction of lymphatics may cause chylous effusions of the thorax and abdomen. Microscopic examination of involved lymph nodes reveals interlacing bundles of smooth muscle cells with intervening sinuses. Similarly, in thoracic duct and retroperitoneal involvement, anastomosing cords of cells enclosing lined spaces are seen.
LAM nodules line distal airways, leading to airway inflammation, air-trapping, and bullae and pneumothorax, perhaps by a check-valve mechanism. It has been suggested that proteolytic enzymes (matrix metalloproteinases) released by the LAM cells play a role in the formation of these cysts, whereas there is no evidence for an imbalance in the α 1 -antitrypsin system. The thin-walled cysts contain foci of LAM nodules covered by hyperplastic type II cells and patches of bronchiolar epithelium. Focal proliferations of type II pneumocytes (multifocal micronodular pneumocyte hyperplasia [MMPH]) may occur, almost exclusively, in patients affected by TSC. MMPH has been reported, however, in men and women with and without TSC, in men and women with TSC and LAM, and in women with S-LAM. MMPH as a histologic lesion does not seem to be clinically important, although there is one case report of a patient without underlying TSC or S-LAM who died of respiratory failure ascribed to MMPH.
Besides MMPH, the spectrum of pulmonary lesions associated with S-LAM and TSC continues to broaden and now comprises AML, focal clear cell tumors, and micronodular/interstitial proliferation of clear cells, atypical adenomatous hyperplasia, and lepidic adenocarcinoma. In addition, there is an assortment of abdominal lesions, many TSC-related, characterized by the proliferation of LAM cells. These include AML and its morphologic variants (leiomyoma-like AML, lipoma-like AML, oncocytoma-like AML, monotypic epithelioid AML), extrapulmonary clear cell “sugar” tumors, lymphangioleiomyoma, and renal capsuloma.
Lung Function
Normal spirometry may be present in nearly one-third of patients with S-LAM and in half of patients with TSC-LAM. The most frequent abnormalities of pulmonary function in patients with LAM are airflow obstruction, characterized by a reduction in forced expiratory volume in 1 second (FEV1) and the FEV1-to–forced vital capacity ratio, and decreased diffusion capacity for carbon monoxide. Abnormalities of gas transfer and FEV1 do not parallel each other, however. Fixed and reversible airflow obstruction is thought to be caused by airway dysfunction more than by loss of lung elastic recoil, whereas decreased diffusion capacity for carbon monoxide probably reflects loss of gas exchange area because it is most closely paralleled by the LAM histologic score.
Restriction or combined restriction and obstruction are frequent functional deficits, particularly in patients with history of pleurodesis. Increased total lung capacity and an increased residual volume–to–total lung capacity ratio also are frequent. Hypoxemia occurs even in patients with near-normal diffusion capacity for carbon monoxide and FEV1. Cardiopulmonary exercise testing uncovers the presence of exercise-induced hypoxemia and assists in grading the severity of disease and may be helpful in determining supplemental oxygen requirements in patients with LAM.
Manifestations of the Disease
Radiography
The most common radiographic abnormality is a widespread fine reticular or reticulonodular pattern, reflecting the superimposition of the thin-walled cysts ( Fig. 35.5 ). The chest radiograph is often normal in early disease and in up to 26% of symptomatic patients. In general, radiographic abnormalities are subtle and delicate but correlate roughly with disease severity, with more apparent and coarser reticulation and cystic changes in patients with more advanced pulmonary involvement. The reticular pattern is less coarse and prominent than the honeycombing of end-stage interstitial fibrosis. Sometimes linear opacities, analogous to Kerley B lines and representing dilated or obstructed lymphatic channels, can be identified. Ill-defined or ground-glass opacities may reflect pulmonary hemorrhage, edema, or hyperperfusion of normal residual lung parenchyma. Large nodules and miliary opacities have been described; some of them likely reflect MMPH in patients affected by TSC. Solitary nodules or masses also may represent clear cell sugar tumor, pulmonary AML, or atelectasis.

Patients with S-LAM and patients with TSC-LAM have similar frequencies of radiographic abnormalities. Perihilar opacities resulting from pulmonary artery aneurysms and patchy bony sclerosis in the ribs and spine are uncommon manifestations described only in patients with TSC. Hilar and mediastinal lymphadenopathy is not a radiographic feature of LAM.
The distribution of the interstitial opacities tends to be symmetric and diffuse, although a slight predominance at the lower lung zones may be observed. Lung volumes are normal initially but tend to increase over time, likely because of small airways disease, air-trapping, and cyst formation. The combination of delicate reticular opacities and normal or expanded lungs is typical of LAM. The chest radiograph may reveal an unsuspected pneumothorax, with or without underlying parenchymal opacities (see Figs. 35.1 and 35.2 ). The incidence and recurrence rates of secondary spontaneous pneumothorax in LAM are higher than in other chronic pulmonary disorders. LAM is a recognized cause of bilateral pneumothoraces and should be a primary differential diagnosis in a woman of childbearing age presenting with bilateral pneumothoraces without an obvious explanation. Pleural effusion, typically chylothorax, is another characteristic finding in patients with LAM. The pleural effusions are more often unilateral than bilateral and tend to be large and recurrent (see Fig. 35.2 ).
Computed Tomography
Pulmonary cysts are the cardinal CT feature of LAM. CT may show cysts even when the chest radiograph appears normal or may reveal a radiographically occult pleural effusion or pneumothorax (see Fig. 35.1 ). Thin-section CT is more sensitive than 5-mm-thick section CT at detecting cysts and assessing their extent and distribution. The minimum-intensity projection algorithm may be useful for improving visualization and assessment extent of the LAM cysts ( Fig. 35.6 ).

The CT appearance of pulmonary LAM is often striking and characteristic ( Figs. 35.6 to 35.8 ; see Fig. 35.1 ). The first challenging step for radiologists is to identify pulmonary changes confidently as cysts per se, particularly because the CT appearances can be similar to centrilobular emphysema as discussed later ( Fig. 35.9 ). Cysts are distributed equally and symmetrically throughout the central and peripheral lung parenchyma ( Fig. 35.10 ; see Figs. 35.6 to 35.8 ). Either relative apical or basal sparing has been reported, however, and a focal unilateral disease has been described in a patient with TSC-LAM. However, in the early stages of the disease, cysts are small, few, and scattered. The European Respiratory Society guidelines for the diagnosis and management of LAM consider the CT features, respectively, as characteristic of LAM when the cysts’ number is greater than 10, and as compatible with LAM when greater than 2 and less than or equal to 10. Lung cysts are generally more extensive in S-LAM than in TSC-LAM (see Table 35.1 ).