3 Malformations of the Spinal Canal
D. Uhlenbrock and D. Brechtelsbauer
Contents
Embryogenesis and Maldevelopments
Development of the Central Nervous System
The Caudal Differentiation and Regression Phase
Formation of the Vertebral Column
Theories of the Pathogenesis of Myelomeningoceles and Arnold-Chiari Type II Malformation
Theories of Pathogenesis of Closed Spinal Malformations
Myelocele and Myelomeningocele
Intradural Lipoma and Lipomyelomeningocele
Fibrolipomatosis of the Filum Terminale
Split Cord Malformation Types I and II
Partial and Total Sacral Agenesis
Arnold-Chiari Type I Malformation
Arnold-Chiari Type II Malformation
Non-communicating Syringomyelia
The majority of cases concern dysraphic malformations, although the slightly less common non-dysraphic malformations can also lead to disorders resulting from malformations of the midline. They too are classified under malformations of the spinal canal and will therefore also be dealt with here. Table 3.1 provides an overview of the malformations.
Definition
Spinal dysraphism. Spinal dysraphism refers to disorders arising from a faulty development of the spine and spinal canal. Specifically they concern pathological connections of varying degrees of expression between bony structures, neuronal tissue and the subcutis or cutis. The spine itself usually exhibits disturbances of fusion as well as bony defects. Spinal dysraphism is classified into:
• open (spina bifida aperta) forms,
• closed (occult spinal dysraphism) forms.
Dysraphic malformations:
Open spinal malformations • myelocele • myelomeningocele • myelomeningocystocele Closed spinal malformations • meningocele • spinal lipoma • lipomyelomeningocele • fibrolipomatosis of the filum terminal • tethered cord syndrome • split cord malformation types I and II • dermal sinus • split notochord syndrome
Non-dysraphic malformations:
• sacral agenesis • congenital tumors (teratoma, dermoid, epidermoid) • spinal cysts • Arnold-Chiari types I and II malformations • syringomyelia • congenital spinal stenosis |
Open spinal malformation (spina bifida aperta): This involves myelomeningocele (MMC), including its subforms (myeloschisis, hemimyelomeningocele, syringomyelomeningocele) and myelomeningocystocele.
The term “open” is supposed to express the fact that parts of the spinal canal are displaced dorsally through a bony defect of the spine and reach the level of the skin or protrude above skin level. There is direct contact between the spinal cord or the meninges or both, and the skin resulting in impending or manifest exposure of the spinal canal to the exterior.
Thus, in the presence of myelomeningocele, the spinal cord is exposed at the level of the skin and presents a direct risk of infection. Surgical intervention is therefore necessary during the first 24–48 hours after birth. After this time it is not uncommon for watchful waiting to lead to a rapid deterioration of the neurostatus. Such children also often present with hydrocephalus (Arnold-Chiari type II malformation), which will require shunt insertion after birth.
Closed spinal malformation (occult spinal dysraphism): A whole spectrum of clinical pictures are classified under this term whose common feature is that defects of the spinal canal are covered by skin and subcutaneous tissue. The neuronal structures do not herniate above skin level. When elevations do appear in the region of the back then they are characteristically caused by a large subcutaneous lipoma. At the level of the defect, however, skin changes may present in the form of hypertrichosis, pigment disturbances, angiomas, dimples or fistula tracts. In occult spinal dysraphism too, individual cases will present alterations in the form and function of the spinal cord with varying degrees of expression.
Spina bifida occulta. This merely refers to an incomplete fusion of the dorsal vertebral elements, especially the vertebral arch, without any changes within the spinal canal. The finding therefore bears no clinical significance.
Important information for the radiologist
Open spinal malformations are not normally examined using imaging techniques but undergo immediate surgery. This is done with the idea of preventing infections of the spinal canal at all events. Imaging techniques are not usually used until the open malformations have been repaired and clinical problems arise after surgery. Operations on closed spinal malformations on the other hand depend on the clinical picture. Here, there is usually no urgency, with the exception of a dermal sinus where surgery of this disorder is also done swiftly to avoid infections.
So in the other cases there remains enough time for an extensive and thorough examination, primarily using MRI and only seldom is myelography or CT myelography required. The latter are used above all for spinal cysts where the question of a communication between the subarachnoid space and the contents of the cyst can only be answered myelographically.
Understanding the pathogenesis of dysraphic malformations can only be gained with knowledge of their embryogenesis. The following chapter presents the embryogenesis in great detail and includes the various theories on the malformations of myelomeningocele and the Arnold-Chiari types.
Embryogenesis and Maldevelopments
Knowledge of embryogenesis and possible maldevelopments facilitates the understanding of congenital anomalies of the spine and spinal canal. The malformations can be explained by certain developmental disturbances occurring at various times during embryogenesis.
Development of the Central Nervous System
The development of the CNS is divided into different phases:
• the neurulation phase,
• the phase of caudal differentiation (canalization) and regression.
 The Neurulation Phase
The Neurulation Phase
The neurulation phase covers the period of the development of the neural plate and the neural tube until their proximal and distal closure. This is reached by day 28 of embryonic development. After conception, a flat oval disk forms by cell division and is fully differentiated within 14 days into the two tissue layers—the ectoderm and the endoderm. On this disk, at its caudal end, ectodermal cells accumulate longitudinally to form a plate, the so-called primitive streak. At the cranial end of this streak a round pit forms, and around this pit cells proliferate to form a peripheral wall called Hensen’s node (Fig. 3.1).
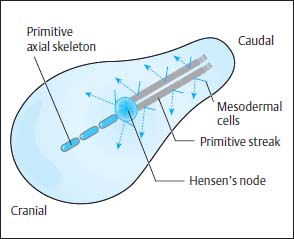
Fig. 3.1 Embryonic development before the onset of the neurulation phase. Cells migrate from the primitive streak to form the mesoderm. The primitive axial skeleton is formed from the cells emanating from Hensen’s node.
Along the primitive streak cells enter the disk and spread out in a cranial direction between ectoderm and endoderm to form the mesoderm. In addition, on days 15 and 16, cells migrate inward via Hensen’s node and spread out cranially in the midline to form the primitive axial skeleton, the notochordal process (Fig. 3.1). The notochordal process develops a centrally open canal with a connection to the exterior via the primitive pit (Fig. 3.2).
The neurulation phase begins around 16–17 days after conception with the development of the neural plate. This process is induced by the notochordal process and begins with a thickening of the ectoderm along the notochordal process, cranial to Hensen’s node. Formation of the neural groove occurs along the length of the notochordal process, the lateral margins of which thicken (Fig. 3.3).
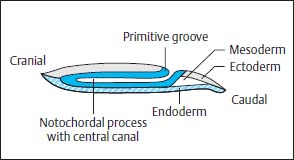
Fig. 3.2 Notochordal process with central canal.

Fig. 3.3 Development of the neural plate at the beginning of the neurulation phase. The neural plate initially displays only a slight indentation. The notochordal process lies separated from both the endoderm and the ectoderm.
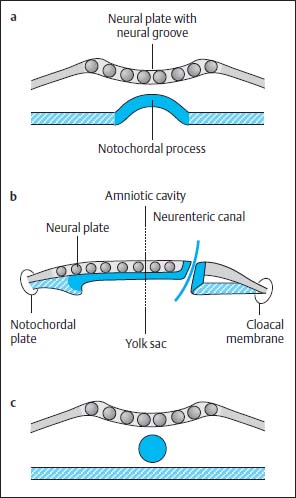
Fig. 3.4a–c Notochordal process:
a The notochordal process embedding into the endoderm. The central canal degenerates, while the notochordal process forms a longitudinal groove. The neural groove displays a more marked indentation.
b Temporary communication between the amnion cavity and yolk sac.
c Division of the notochord from the endoderm and formation of a cylinder.
At this time the notochordal process is reorganized to form a plate, which is embedded into the endoderm. The central canal of the primitive axial skeleton thus degenerates and in its place an elongated groove develops in the midline. The primitive axial skeleton gains contact with the yolk sac by incorporation into the endoderm (Fig. 3.4 a).
At the caudal end of the plate where the primitive groove merges into the axial skeleton, a communication now develops between the yolk sac and the amniotic cavity, the so-called neurenteric canal (Fig. 3.4b).
This canal degenerates again after 2–6 days due to separation of the notochordal plate from the endoderm, the endoderm once again forming a complete continuous covering and the cells of the primitive axial skeleton gathering to form a cylinder in the midline, the definitive notochord (Fig. 3.4c). Meanwhile the neural plate is further transformed. The neural groove deepens. The elevations on either side of the neural plate develop into the future neural crest. The neural plate is reorganized to form the neural tube by dorsal closure of the U-shaped plate, separation from the ectoderm and formation of an intact ectodermal surface, the future skin. Mesodermal tissue is formed between the ectoderm and the neural tube so that there is no longer any direct contact between the two tissues. The neural cells at either margin of the neural plate migrate downwards to accumulate dorsal to the neural tube where they form the neural crest (Fig. 3.5).
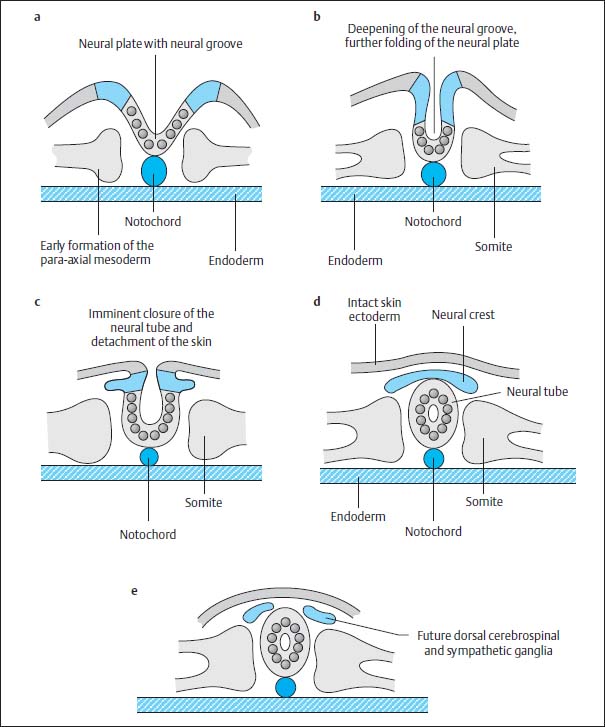
Fig. 3.5a–e Development of the neural tube by fusion of the neural primordium.
At the same time mesodermal cells are transformed paraxially on either side of both the notochord and the neural tube to become the so-called primitive segments, the future somites, which flank the axial skeleton from around day 16–17. After 28 days, 30 of these original segments have already formed. They differentiate dorsomedially into the myotomes and eventually into the segmental paraspinal muscles. The anteromedial components develop into the sclerotomes and eventually into vertebral bodies, vertebral arches, ligaments and meninges, as well as into the ribs and intercostal muscles. The anterolateral elements develop into the dermatomes and form the skin, subcutaneous tissue and the muscles of the abdominal wall (Fig. 3.6).
The process of fusion of the neural plate begins in the mid portion and is continued in a caudal and cranial direction. The rostral end of the neural tube closes on day 24, the caudal end on days 26–28. The exact site of the caudal end is disputed, but most likely it is located in the distal lumbar region (Fig. 3.7).
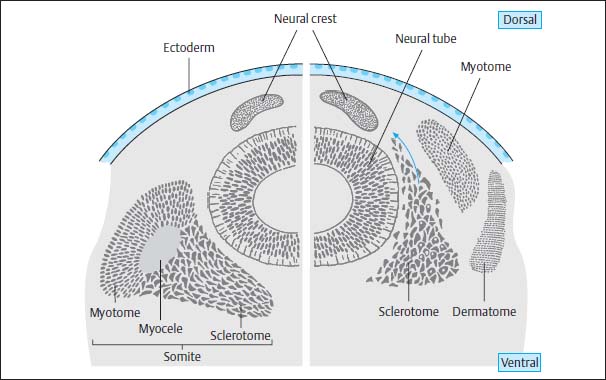
Fig. 3.6 Diagram of the formation of the somites with subsequent division into myotome, sclerotome, and dermatome. The sclerotome itself divides into three major groups of mesodermal cells: the dorsal group forms the vertebral arches and the primitive meninges, the medial group the vertebral bodies, and the ventral group the ribs and intercostal muscles.
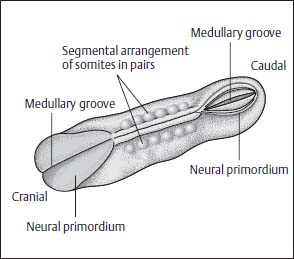
Fig. 3.7 Somites. The neural primordium closes first in the mid portions. The somites are shown arranged in pairs along the midline.
Hensen’s node and the primitive streak then undergo regression and migrate in a caudal direction to the future coccygeal region.
 The Caudal Differentiation and Regression Phase
The Caudal Differentiation and Regression Phase
The formation of the spinal cord begins caudal to L4 with the cells of Hensen’s node and of the primitive streak forming an undifferentiated cell mass beneath the intact ectodermal surface after their migration caudalward. Within this accumulation of cells, vacuoles are formed which coalesce, consequently leading to the formation of a tube surrounded by ependyma-like cells. This cell accumulation and its inner canal fuse with the neural tube (canalization). Around day 38 of embryonic development regression of this lumbosacral canal begins. The cell accumulation and the inner canal diminish in size, around days 44–48 cell necrosis occurs and the formation of the conus, ventriculus terminalis and filum terminale takes place. The filum is attached to the fifth coccygeal vertebra as a ligament. The vertebral column and the spinal cord grow at different rates resulting in the ascension of the conus medullaris. In the fourth week the conus is at about the L4/L5 level, in the second week at L3/L4, in the 40th week at L2/L3, and 10 weeks later reaches the adult level (L1/L2) (Barson, 1970; Di-Pietro, 1993).
Development of the Meninges
The meninges develop immediately after the formation of the neural tube, particularly after completion of its distal end. The caudal end of the dural sac reaches down to about S4 in the fetus and is situated at the S2 level in the adult. Thus the dural sac is subject to a less pronounced ascension compared with the conus medullaris.
Formation of the Vertebral Column
The vertebrae develop from the sclerotome cells, with one vertebra in each case being formed from the cells of the next higher and lower sclerotome. In addition, the cells of the right and left side merge to form one vertebra so that in total four groups of sclerotome cells are involved in the formation of one vertebra. The sclerotome cells lying between the vertebral bodies develop into the annulus fibrosus; the notochord is transformed into the nucleus pulposus. The notochord in the vertebral body undergoes complete regression. The dorsal components of the vertebrae are formed from paired sclerotome cells of the right and left side. The lumbosacral vertebrae develop in a less orderly fashion than the proximal vertebrae. Cells of the axial skeleton, the mesoderm and the caudal neural tube are transformed into somites (primitive segments), which in turn form the sacral and coccygeal vertebrae, and subsequently are then subject to regression, fusion and transformation. During this process a series of anomalies can develop, such as:
• lumbosacral lipoma,
• teratoma,
• sacral agenesis.
Pathogenesis of Anomalies
Theories of the Pathogenesis of Myelomeningoceles and Arnold-Chiari Type II Malformation
There are a number of theories concerning the development of myelomeningoceles and Arnold-Chiari type II malformations, which will be outlined below. The individual ideas emanate in part from very different standpoints. For example, the question of whether spinal dysraphism develops before the completion of the neural plate and skin or afterwards is still disputed. The fact that myelomeningocele or myeloschisis is found only in the lumbosacral region might falsely suggest that, because the spinal cord and spine develop in this segment by canalization and regression beneath an intact ectoderm, this malformation usually develops underneath the intact skin which subsequently opens up.
 Theory of Disturbed Cerebrospinal Fluid Circulation
Theory of Disturbed Cerebrospinal Fluid Circulation
Chiari (1891, 1895) and, later, Gardner (1973) considered the cause to lie in a disturbance of cerebrospinal fluid (CSF) circulation in the posterior cranial fossa. In the presence of maldevelopment of the CSF-containing spaces, CSF cannot drain sufficiently from the ventricles and disperses caudally via the neural tube. This results in dilation and disruption of the neural tube and consequently in the development of myeloschisis or myelomeningocele. According to this theory, Arnold-Chiari type II malformation develops from excessive pulsatile pressure of the supratentorial choroid plexus with a corresponding downward displacement of structures in the posterior fossa into the spinal canal.
Objections to this theory have been raised which point out that myeloschisis is already found before the development of the choroid plexus. Furthermore, this theory does not explain the small size of the posterior fossa, the upward herniation of the contents of the posterior fossa, the often slit-like formation of the fourth ventricle, and various other cerebral anomalies.
 Traction Theory
Traction Theory
According to this theory developed by Lichtenstein (1940, 1942) and Penfield and Coburn (1938), the Arnold-Chiari type II malformation is supposed to develop from traction occurring as a result of adherence of the spinal cord during myelomeningocele development. But this theory cannot explain why the dorsal components of the pons and medulla oblongata are mainly involved and the ventral elements less so. Traction does not explain the dorsal Z-shaped angulation of the medulla oblongata, so there must be pressure from above. Furthermore, myelomeningoceles also occur without Arnold-Chiari type II malformation, and Arnold-Chiari type II malformations are never present in occult spinal dysraphism despite tethering of the conus.
 Theory of Maldevelopment of Segments of the Brain Stem
Theory of Maldevelopment of Segments of the Brain Stem
Cleland (1883) primarily suspected maldevelopment of the brain stem as the cause of the malformation. This theory was further developed by Daniel and Strich (1958) and Peach (1965) who added that an insufficient or absent angulation of the pons, in particular, would result in maldevelopment with corresponding upward or downward displacement of parts of the brain. The question of why the angulation of the pons is absent remains unanswered, and also of how the other cerebral changes are to be explained.
 Theory of Overgrowth or Underdevelopment of the Posterior Fossa
Theory of Overgrowth or Underdevelopment of the Posterior Fossa
Based on the results of animal studies, Padget (1968, 1970, 1972) supported the theory of a reopening of the closed spinal cord in the course of the development of spina bifida aperta. According to this theory, fluid is supposed to collect at circumscribed sites within the closed neural tube and, through pressure, forces an opening in the myelon and the skin or, as in anterior spina bifida, contributes to the formation of a ventral cleft. The pathogenesis of Arnold-Chiari malformation is said to be associated with the development of the neural malformation, inasmuch as microcephaly and, consequently, reduction in size of the posterior fossa occur as a result of fluid escape at the level of the spina bifida aperta. The subsequent development of the cerebellum within the restricted posterior fossa is supposed to result in a downward displacement of parts of the midbrain and cerebellum. The theory of an embryonic microcranium of the posterior fossa as a cause of the Arnold-Chiari type II malformation gains support from the fact that, despite a pressure-producing hydrocephalus, these patients in fact demonstrate only a normal-sized skull and never develop a microcranium after shunt insertion.
Marin-Padilla and Marin-Padilla (1981) reached similar conclusions on the basis of results of animal studies. An underdevelopment of the occipital bone, especially the basal parts, was achieved by treating hamster embryos with vitamin A, which resulted in an insufficient development in the size of the posterior fossa. The fossa was unable to contain the cerebellum and brain stem within the predefined bony coverings so that parts were displaced caudally, toward the foramen magnum, yet were never seen within the cervical canal. For this reason, and due to the fact that further malformations associated with Arnold-Chiari malformation are not amenable to explanation by this theory, this concept has been regarded as unsatisfactory.
Theories originating from Barry (1957), and in a similar form from Brocklehurst (1969, 1971), also assume a disproportion between bony coverings and neuroepithelium in the posterior fossa. Barry suspected an excessive growth of neuroepithelial structures in the posterior fossa as a factor, while Brocklehurst assumed a disproportionate development of neural ectoderm and investing mesoderm.
 Currently Accepted Theory
Currently Accepted Theory
The theory that has enjoyed increasing acceptance over recent years was expounded by McLone and Knepper (1989) and is based on continuing animal studies. It differs only partly from the results obtained by Padget and Marin-Padilla in particular, while largely reaching similar conclusions. The animal studies yielded the result that the development of myelomeningocele, Arnold-Chiari malformation and the other cerebral malformations must be ascribed to a uniform pathogenesis according to which the regulation of their development is disturbed primarily on an enzymatic level—a development that is normally genome-induced and based on intra- and extracellular enzymatic processes. The starting point is the incomplete closure of the neural crest which, due to loss of CSF, leads to the collapse of the roof of the hindbrain (rhombencephalon) and thus to an inadequate expansion of the primitive ventricles. This results in an insufficient expansion of the posterior fossa. The development of the cerebellum and brain stem continues in the presence of the predetermined, yet insufficient, form of the bony coverings of the posterior fossa. The junction between brain stem and spinal cord is displaced below the foramen magnum. The angle of the brain stem is altered, parts of the cerebellum and the brain stem move cranially and caudally. A faulty development of the nuclei of the cerebral nerves results from this. The primitive ventricles become insufficiently filled with fluid, which results in disorganization of the cerebral cortex. The mechanical support of the primitive ventricles is a precondition for the normal organization of the cortex. The insufficient development of the cerebral hemispheres results in microgyria and ectopy as well as in dysgenesis of the corpus callosum. The third ventricle remains small, which prevents the separation of the thalamic nuclei and leads to the formation of the intermediate mass. The inadequate expansion of the brain results in a disorganization of the collagen bundles of the cranial vault. Instead of radial lines the collagen forms winding and rounded collagen bundles, which are associated with the characteristic development of the lacunar skull (craniolacunia or Lückenschädel).
The hydrocephalus often associated with myelomeningocele can have various causes. On the one hand there may be a blockage of the basal cisterns and the subarachnoid space due to the downward displacement of parts of the cerebellum and brain stem or, alternatively, an occlusion of the aqueduct or of the foramina of the fourth ventricle can be the cause. Not least, a disturbance of circulation may be present, resulting from a blockage at the level of the tentorium. At the time of birth a large number of children present with microcrania that rapidly regress after closure of the sac (cele), so that the head circumference increases. With limited growth capacity of the bony coverings, hydrocephalus usually develops together with the increase in volume of the posterior fossa, which accompanies brain tissue growth. This leads to a blockage of the CSF pathways, particularly the CSF circulation at the level of the tentorium. Thus the development of hydrocephalus is also the result of the disproportionate growth of the cerebral structures and the cranial vault.
Theories of Pathogenesis of Closed Spinal Malformations
Pathogenesis of closed spinal malformations can arise from five different causes:
• failure of various tissues to separate,
• malformation of the neural tube,
• faulty development of the primitive axial skeleton,
• faulty development of the neurenteric canal,
• errors in canalization and regression at a lumbosacral level.
 Lipoma and Lipomyelomeningocele
Lipoma and Lipomyelomeningocele
As a rule, the separation of ectoderm and neural tissue is a well-regulated process that proceeds in a bilaterally equal manner and during which the closure of the neural tube happens at the same time as the mesenchyme develops between the ectoderm and neural tube. Here the mesenchyme has no contact with the central canal. If the separation between ectoderm and neural tissue occurs prematurely or unilaterally, a disturbance of mesenchymal architecture can occur, possibly resulting in contact between mesenchymal cells and the central canal before the closure of the neural primordium.
Apparently mesenchymal tissue that gains contact with the neural groove and the future central canal is transformed into fat. This explains the development of lipoma and lipomyelomeningocele along the course of the spinal canal with the exception of the lumbosacral segments (Caram et al., 1957; Emery and Lendon, 1969; Anderson, 1975; Bruce and Schut, 1979). In the lumbosacral region the pathogenesis can be explained by a faulty canalization and regression, inasmuch as the cell-tissue differentiation into a neural and mesenchymal component is inadequate and mesenchymal structures become enclosed in the spinal canal.
 Dermal Sinus
Dermal Sinus
The dermal sinus can be explained by absent or insufficient separation of ectodermal from neural tissue, resulting in a tract that connects the skin with the neural structures within the spinal canal. Epidermoids and dermoids can also arise from the ectodermal tissue and are often associated with a dermal sinus (Walker and Bucy, 1934).
 Tethered Cord Syndrome
Tethered Cord Syndrome
The process of canalization proceeds normally in the case of tethered cord syndrome, but errors of regression occur that consequently result in alterations of the filum terminale, such as fibrolipomatosis, a thickening or shortening, and in some cases the formation of connective-tissue bands that tether the spinal cord to the dura. This results in the fixed low-lying conus during the ascension phase (Jones and Love, 1956).
 Split Cord Malformation, Neurenteric Cyst, Combined Anterior and Posterior Spina Bifida
Split Cord Malformation, Neurenteric Cyst, Combined Anterior and Posterior Spina Bifida
These changes, which predominantly affect the thoracic and cervical regions and are frequently associated with malformations of the vertebrae, can be regarded as different expressions of a uniform process of malformation.
During embryonic development, a canal is occasionally formed that connects the amnion cavity with the yolk sac. This canal only exists for a few days and then closes again. The cause of the malformation is not considered to be insufficient regression of this canal, however, but rather that an accessory neurenteric canal develops above the normal physiological canal (Pang et al., 1992).
This accessory canal also develops in the midline and represents a communication between yolk sac and amnion cavity. It leads to a splitting of the neural plate and notochordal process, interfering with the development of the vertebrae and the neural tube in this segment.
The assumption that an accessory canal proximal to the proper physiological canal is the cause of the respective malformations is based on the fact that the physiological canal lies at the level of Hensen’s node and during the caudal migration of Hensen’s node comes to lie at the level of the coccyx. The resulting malformations, however, are always found above the coccygeal region.
The accessory canal is additionally surrounded by mesenchyme so that an endomesenchymal tract develops along the canal and occupies the space between the split notochordal process and the divided neural plate.
The final degree of expression of the malformation depends on three factors:
• the ability of the embryo to maintain the healing process along the endomesenchymal tract,
• the ultimately variable extent of the endomesenchymal tract,
• the final and definite development of the displaced mesenchyme and endoderm along the midline.
The split notochordal process is more able than the divided neural plate to initiate a process of healing along the fistula, resulting in the finding that clefting of the spinal cord is more often encountered than split vertebrae.
Although the entire endomesenchymal tract connecting bowel and skin can persist, it is more common for only parts of the tract to remain.
The endoderm can degenerate. Alternatively, it can form cysts lined with respiratory or gastrointestinal epithelium, both of which are derived from the endoderm.
Because the pluripotent mesenchyme is capable of transforming itself into fibroblasts, cartilaginous or bone cells as well as blood vessels, fat and myoblasts, the sagittal septum dividing the spinal cord can be composed of various tissues.
If the dorsal component of the endomesenchymal tract degenerates and the ventral portion persists, then this structure can connect the bowel with parts of the vertebra or spinal cord.
Inside this tract the endoderm can differentiate into intestinal epithelium, leading to an intestinal duplication that is connected to a vertebra or the spinal cord.
The development of this band can come to a standstill if no further endodermal differentiation takes place and can sometimes be the cause of intestinal malrotation inasmuch as the anticlockwise rotation of the bowel is prevented (McLetchie et al., 1954; Beardmore and Wiglesworth, 1958; Bentley and Smith, 1960; Pang et al., 1992).
Dysraphic Malformations
Open spinal Malformations
 Myelocele and Myelomeningocele
Myelocele and Myelomeningocele
Definition
In myelocele and myelomeningocele the spinal cord is exposed at a circumscribed site so that there is neither a bony nor cutaneous covering.
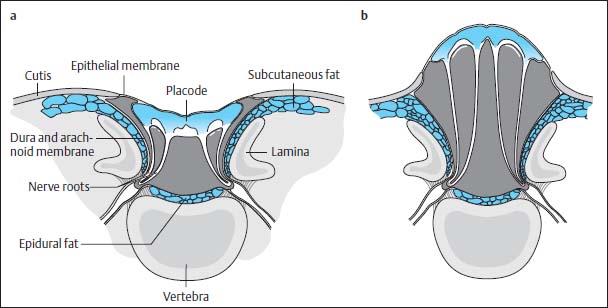
Fig. 3.8a, b Myelocele and myelomeningocele.
a Myelocele: The neural placode is a flat plate of neural tissue lying directly level with the skin. Dorsally the dura is non-existent. The pia mater and arachnoid membrane border on the ventral surface of the placode and dura, forming an arachnoid pocket or cyst, which is connected to the subarachnoid space proximally and distally. Both the dorsal and the ventral nerve roots arise from the ventral surface of the placode.
b Myelomeningocele: In contrast to myelocele, there is an expansion of the ventral subarachnoid space dorsally leading to a bulging protrusion of the placode above skin level.
The spinal cord usually appears flat and stretched out (placode) and lies on a level with the skin.
The sac (cele) can extend far dorsally (myelomeningocele) or can lie almost on a level with the spinal canal (myelocele) (Fig. 3.8).
Morphology
The skin generally forms the margin of the placode because in this type of malformation the neuroectoderm does not separate from the skin. Since the mesenchymal structures had no chance of occupying the region between skin and neural tissue they are not located dorsal to the neuroectoderm but are found anterolateral to the neural tissue. The skin is attached to the placode by a narrow strip of leptomeninx, which in turn is covered by epithelium (epithelial zone). The dura and arachnoid membrane do not invest the placode. Instead the meninges develop exclusively ventral to the placode.
The dura is contiguous with the skin on the lateral margin of the epithelial zone with the arachnoid membrane contributing to the formation of the epithelial zone.
The placode represents incompletely formed spinal cord in the sense that the lateral ends of the embryonic neural plate have not joined together. Consequently all the dorsal nerves enter and exit ventrally with the dorsal nerve roots lying lateral to the ventral roots. A central canal is either not recognizable at all or is incompletely formed and opens outwards so that CSF can escape freely.
The myelomeningocele is usually associated with severe bony defects. The dorsal vertebral components do not fuse, but instead the pedicles and laminae are displaced laterally and anterolaterally. This results in a distinct enlargement of the spinal canal. The craniocaudal dimensions of the bony defect extend beyond the skin defect. If the bony defect is situated at the level of the thoracolumbar junction, then a complete spina bifida develops from this point caudally and involves the whole lumbosacral space, regardless of the level at which the skin defect is located. At other sites the bony defect remains localized (Fig. 3.9).
The vertebrae can also have defects. Hemivertebrae, aplasias or wedge vertebrae are seen, as well as segmented block vertebrae, which are not infrequently encountered unilaterally. There are also malformations involving the ribs.
In the lumbar region, furthermore, ventral or ventrolateral displacements of the back muscles, including the psoas muscle, can be found. The paraspinal muscles subsequently act as flexors and not extensors. Together with the other bony malformations, this will result in the development of scoliosis.
Location
Myelomeningoceles are located at the following sites (French, 1990):
• 3 % cervical,
• 5 % thoracic,
• 26 % thoracolumbar,
• 25 % lumbar,
• 30 % lumbosacral,
• 10 % sacral.
Pathogenesis
About one-third of patients with myelomeningocele demonstrate an associated type II split cord malformation.
The clefting of the spinal cord can be above (approximately 45 %), below (approximately 30 %) or at the level of (approximately 25 %) the myelomeningocele (Emery and Lendon, 1973). Not infrequently there is also a hemimyelo(meningo)cele of the spinal cord present. It is seen in about 10 % of patients with myelomeningocele. In these cases one part of the spinal cord is developed in the form of a myelomeningocele, while the other part lies in a dural sac separated from the rest of the spinal canal by a fibrous or bony spur (type II split cord malformation). The normal spinal cord can, however, demonstrate tethering by a thickened filum terminale or, in rare cases, is also associated with a myelomeningocele lying caudal to the main finding.
There is also a form of hemimyelomeningocele associated with the occurrence of a type I split cord malformation, i.e. with the presence of a complete and uniform dural sac. In up to 20 % of cases the disorder is combined with an intradural tumor, usually a lipoma, rarely a dermoid or epidermoid.
About 11 % of patients present with syringo(hydro)myelia (Just et al., 1990); in the study population of Emery and Lendon (1972, 1973) the rate was 43 %. Their extent and site vary considerably.
Syringomyelia is found both localized in the cervical region and directly above the myelomeningocele, but also as a complete lesion along the entire length of the spinal cord.
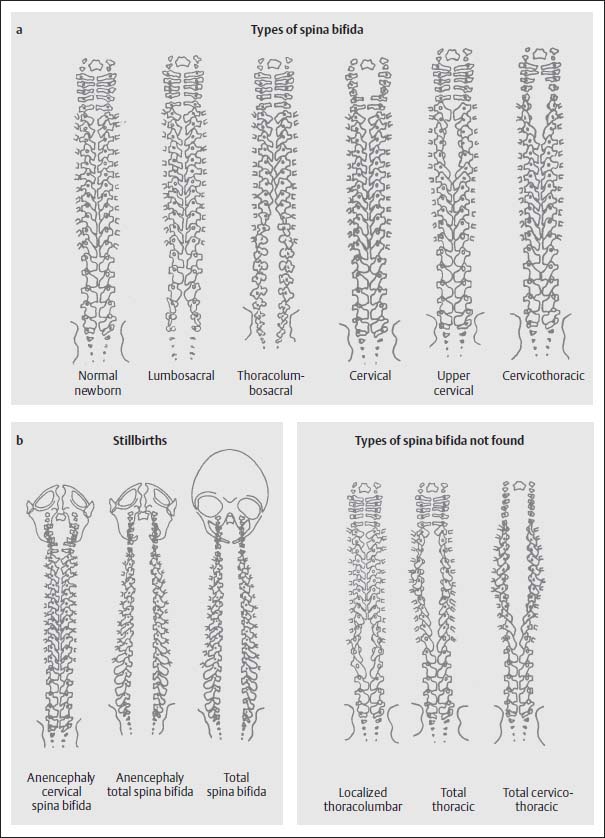
Fig. 3.9a, b Examples of the various types of spina bifida according to location of the defect with involvement of the cranium (from Friede, R.L., Developmental Neuropathology, Springer, Berlin 1989).
Diagnostics
MRI
A preoperative examination of a myelomeningocele using imaging techniques is not usually indicated.
The necessity for an MRI examination only arises when, despite a successful operation, clinical deterioration is noticed after surgery (Figs. 3.10–3.17).
The examination should provide an answer to the following questions:
• Are there any other associated malformations?
• Is there re-tethering after a successful operation?
According to the reports of various authors this is the case in up to 90 % of patients (Just et al., 1990). Admittedly, the diagnosis with the aid of MRI is more a diagnosis of exclusion. Often MRI is unable to demonstrate the offending scar tissue directly. In particular cases it may be wise to perform the examination both in a prone and supine position to detect or exclude dorsal or ventral displacement of an exposed spinal cord.
• Is there an ischemic segment of spinal cord?
An ischemic segment of spinal cord can be well demonstrated by a distinct and circumscribed narrowing of the spinal cord. The cause of ischemia can be due to a technical surgical error. If the ends of the meninges are drawn together too tightly when closing the dural sac, then this can result in narrowing of the dural sac, which keeps the spinal cord relatively constricted, thus compromising circulation.
• Is there a (progressive) syringo(hydro)myelia?
Detection of a syringo(hydro)myelia is important because an untreated syringo(hydro)myelia can contribute to the rapid development of scoliosis. Alternatively, the development of scoliosis in shunted patients can be indicative of shunt dysfunction. (For the pathogenesis of syringo(hydro)myelia see p. 134).
For a better understanding of the surgical management of myelomeningocele, the operative procedure is presented step-by-step (Fig. 3.18).
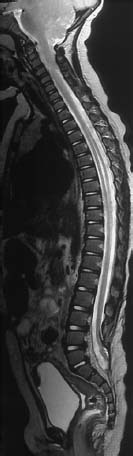
Fig. 3.10 Postoperative lumbosacral MMC. MRI examination using T2-weighting (TSE; TR = 4000 ms, TE = 112 ms) demonstrates the entire spine including the craniocervical junction. There is an Arnold-Chiari type II malformation and a syrinx at the T3–T9 level, with a marked distension of the spinal cord, and spina bifida at the L5/S1 level. The defect has been closed surgically. The spinal cord is elongated as far as S1; at the level of the bony defect there is a dorsal fixation of the conus to the dura (retethering).
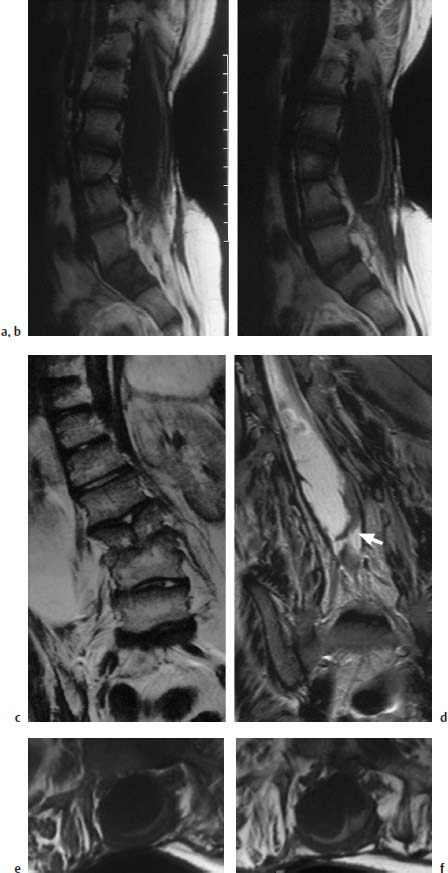
Fig. 3.11a–f Postoperative lumbar MMC.
a, b T1-weighted sagittal section (TSE; TR = 800 ms, TE = 12 ms): The spinal canal is markedly enlarged in a dorsal direction at the level of the long-segment spina bifida. The spinal cord is flat and elongated, displaced dorsally, and exhibits caudal tethering in the lumbosacral region. The dorsal displacement is due to an arachnoid cyst, 8–9 cm in size. Note the vertebral dysplasia at the L3/L4 level.
c T2-weighted coronal section at the level of the split vertebra L3. Marked right convex scoliosis of the thoracic and lumbar spine.
d Slice 1.5 cm further dorsal: the lumbar enlargement of the spinal canal is easily seen together with the fixation of the spinal cord to the dura, particularly on the left (arrow). Apart from this, some band-like scarring is recognizable, which results in an additional fixation of the placode.
e, f Transverse section (SE; TR = 805 ms,TE = 14 ms): The image shows the dorsally very flattened and elongated placode at the level of the spina bifida with a broad fixation in the dural region, particularly towards the left.
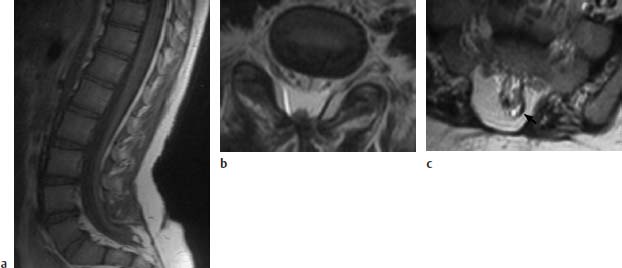
Fig. 3.12a–c Postoperative sacral MMC, now showing retethering:
a T1-weighted sagittal section (TSE; TR = 800 ms, TE = 12 ms): Spinal cord elongated far sacrally and tethered to a small lipoma and the dura at the level of about S2/S3.
b Transverse image using T2-weighting (TSE; TR = 4800 ms, TE = 120 ms): The dorsal tethering of the spinal cord is easily seen on this image. Note the spinal canal projecting slightly dorsally, although no bony interruption is recognizable here.
c Slice 1.5 cm further caudally. In this section the sacral bony interruption is well recognized. The small lipoma dorsolaterally on the left appears signal-intense (arrow). Tethering here of the spinal cord.
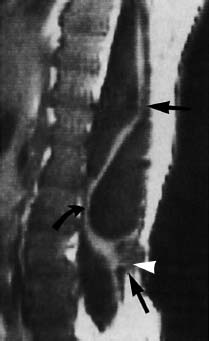
Fig. 3.13 Meningomyelocele. 3-year-old boy with a postoperative meningomyelocele. Note retethering of the spinal cord at two sites dorsally (straight arrow and arrowhead) and ventrally at about the L3 level (curved arrow). A dorsal arachnoidal cyst could be an additional cause of the ventral displacement of the spinal cord (SE; TR = 780 ms, TE = 25 ms).
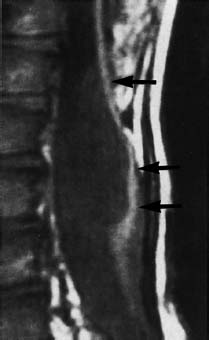
Fig. 3.14 Myelomeningocele. 5-year-old boy with a postoperative myelomeningocele. Note retethering with long-segment dorsal fixation of the spinal cord (arrow) (SE; TR = 780 ms, TE = 25 ms).
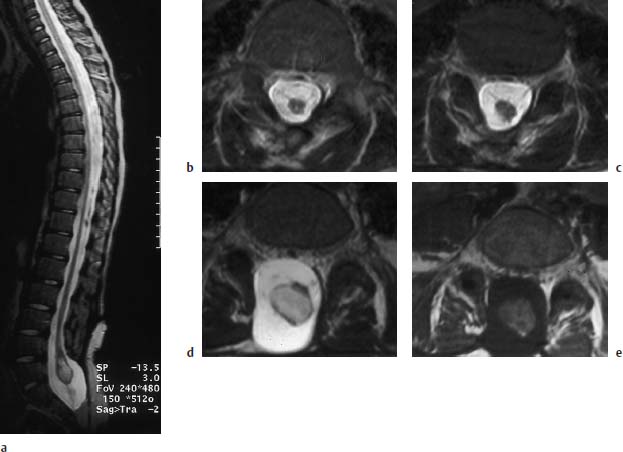
Fig. 3.15a–e Myelomeningocele. 9-year-old boy with a postoperative lumbosacral myelomeningocele. The bony defect is at the L5 level and extends far down sacrally:
a T2-weighted sagittal section (TSE; TR = 5310 ms, TE = 112 ms): Long extended spinal cord reaching as far as L5/S1where it displays a cystic distension at the level of the conus in the form of a ventriculus terminalis. A definite fixation is not recognizable. There is no contact with the dorsal and ventral parts of the dura.
b, c Transverse images (TSE; TR = 5000 ms, TE = 120 ms): Distal parts of the spinal cord with a small punctiform syrinx. The band-like structures traversing in a ventral direction are most likely nerve roots, well recognizable from their alignment with the intervertebral foramina, as in c (5mm caudal to b).
d Slice 15mm further caudal from c. The cystic distension of the conus on this image is well recognizable. Here too there are no definite band-like attachments to the dura.
e Transverse T1-weighted section at the same level as d (SE; TR = 665 ms, TE = 14 ms): The cyst displays fluid with a relatively high signal intensity, most likely as an expression of its protein-rich composition.
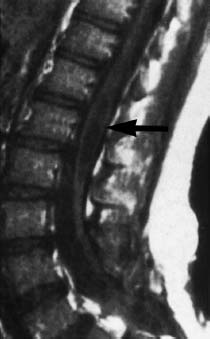
Fig. 3.16 Localized syringo(hydro)myelia above the operative region (arrows). Note retethering of the spinal cord (SE; TR = 720 ms, TE = 20 ms).
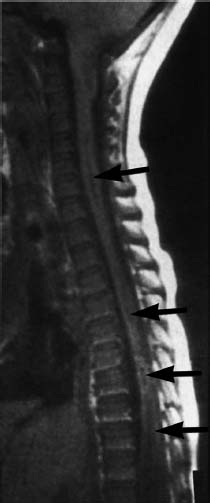
Fig. 3.17 Syrinx formation at multiple sites of the spinal cord (arrow). These regions are not explicitly connected with each other (SE; TR = 650 ms, TE = 25 ms).
 Myelomeningocystocele
Myelomeningocystocele
Definition
The myelomeningocystocele (synonyms: myelomeningohydrocele, syringomyelomeningocele) represents a circumscribed herniation of the widened central canal and dysplastic membranous neural tissue dorsally through a bony defect between the dorsal columns of an otherwise correctly fused spinal cord. The sac (cele) is completely covered with skin.
Location
The finding is mainly seen in the cervical and thoracic regions (Figs. 3.19 and 3.20). The spinal cord can taper off terminally into a cyst into which the often-widened central canal opens (Figs. 3.21 and 3.22). There may be an only slight neurological defect.
Pathogenesis
The disorder is always associated with a caudally or proximally orientated syringomyelia and often with urogenital and anorectal defects.
Bladder exstrophy can develop. In addition scoliosis and partial or complete sacral agenesis are often present.
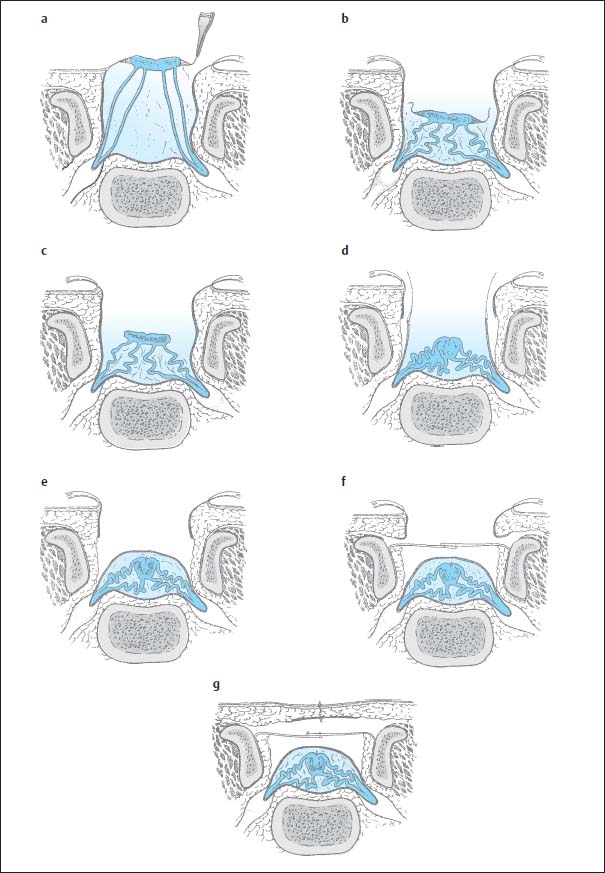
Fig. 3.18a–g Sectional diagrams of operative management of myelomeningocele (from McCullough, D. C. Meningomyelocele. In Raimondi, A.J., M. Shoux, C. Di Rocco The Pediatric Spine, Vol. I, Springer, Berlin, 1989):
a First, the epithelial membrane is detached from the viable normal skin.
b After detachment, the membrane and neural structures are dissected down into the spinal canal.
c The epithelial membrane is dissected off the neural tissue.
d The placode is brought together dorsally and the dura mobilized on either side.
e The dura is closed dorsally.
f The fascia is mobilized, approximated medially, and closed with overlapping sutures.
g Subsequently skin and subcutaneous fat are closed.
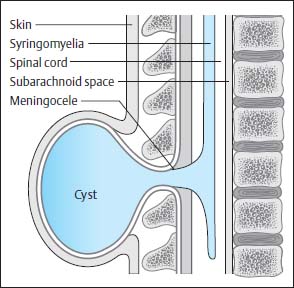
Fig. 3.20 Thoracic myelomeningocystocele. Transverse section of a neonate showing a large sac (cele) formation, which extends far beyond the level of the skin and is connected to the spinal canal via a syrinx.
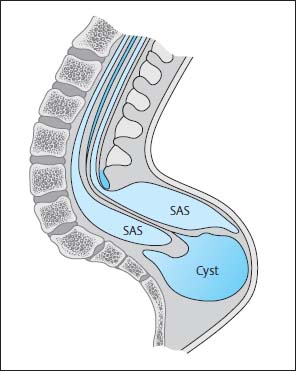
Fig. 3.21 Terminal myelomeningocystocele. There is hydromyelia of the entire spinal cord. The central canal opens into a large cyst at the level of the conus. This cyst lies below the region of the bony spina bifida and, for the most part, below the clearly distended subarachnoid segments that surround the spinal cord distally. SAS Subarachnoid segment
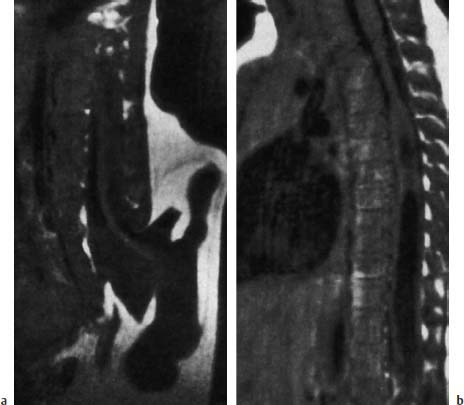
Fig. 3.22a, b Terminal myelomeningocystocele with severe malformations of the urogenital tract and sacral agenesis:
a The spinal cord opens terminally into a large, irregularly bordered cyst that is covered by skin and extensive lipomatous components (SE; TR = 600 ms, TE = 20 ms). Normally cyst and subarachnoid space do not communicate, but this is not always discernible on MRI.
b Severe syringomyelia along the spinal cord (SE; TR = 600 ms, TE = 20 ms).
Closed Spinal Malformations
 Meningocele
Meningocele
Definition
Meningocele consists of dorsal herniation of the dura and arachnoid membrane with a more or less distinct bulging protrusion of the skin.
Location and Pathogenesis
The bony defect tends to be small and can be confined to one vertebra. When more than one vertebra is involved, there is always an enlargement of the spinal canal. The sac (cele) does not usually contain any neural tissue, but in individual cases nerve roots can be found in the sac, especially when the bony defect is very wide. This can lead to tethering of the spinal cord. Arachnoidal adhesions can also lead to secondary fixations and closure of the sac. Normally the meningocele is covered by skin, but pressure necrosis can result in perforations. In this case there is an immediate indication for surgery.
Clinical symptoms, not usually present, are more likely to indicate a myelomeningocystocele.
Meningocele shows a different distribution pattern than that of myelomeningocele (French, 1990):
• 10 % cervical,
• 15 % thoracic,
• 4 % thoracolumbar,
• 37 % lumbar,
• 19 % lumbosacral,
• 15 % sacral.
Diagnostics
MRI
MRI serves the following purposes (Figs. 3.23 and 3.24):
• identification of the lesion,
• exact definition of site and extent,
• verification or exclusion of further changes (e.g. myelomeningocystocele),
• demonstration of the conus medullaris and filum terminale.
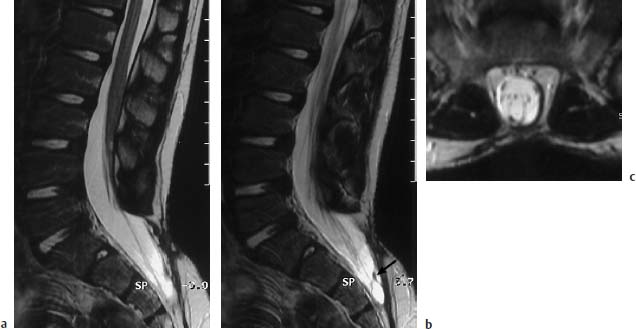
Fig. 3.23a–c Meningocele. 6-year-old boy with postoperative sacral meningocele:
a T2-weighted sagittal section (TSE; TR = 5300 ms, TE = 120 ms): Bony defect at the S2/S3 level dorsally with herniation of the dural sac. Marked cutaneous and subcutaneous scar-tissue formation after surgery. The filum terminale appears band-like, seems shortened, and there is a corresponding low conus at the L3 level.
b Parasagittal section 3mm lateral from a: In the sacral region band-like intradural changes are to be found, which radiate both dorsally and ventrally into the dura. These can be aberrant neural structures, but also are more likely to be scar-tissue changes (arrow).
c T2-weighted transverse section (TSE; TR = 4600 ms, TE = 120 ms): Dorsal filum terminale, possibly tethered to the dura. On this image too, horizontally running structures are noticeable which possibly correspond to aberrant nerve roots. Marked extensive scarring of the skin.
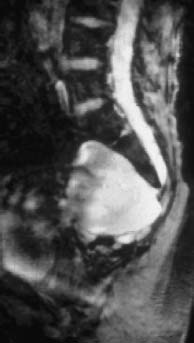
Fig. 3.24 Large anterior sacral meningocele (T2-weighted GRE sequence). Sagittal section showing an anterior meningocele in continuity with the sacral dural sac. Note the correspondingly typical signal iso-intense with CSF in the presacral parts.
 Intradural Lipoma and Lipomyelomeningocele
Intradural Lipoma and Lipomyelomeningocele
Intradural Lipoma
Definition
The intradural lipoma is an accumulation of fatty tissue, which overlies the spinal cord in an extramedullary location, although it is also seen at extra- and intramedullary sites. The lipomas are, however, never completely intramedullary, but only demonstrate an expansion within the central canal, sometimes far cranially and originating from an extramedullary component.
If they are situated at the level of the conus or at a lumbosacral level, they are associated with a tethered cord syndrome and low-lying conus. However, two-thirds of all lipomas found do not demonstrate any spinal cord fixation. The lipomas account for 1 % of all primary intradural tumors (Giuffre, 1966). They are most commonly found at a thoracic level.
Incidence
The intradural lipoma and the lipomyelomeningocele are the most frequent forms of occult spinal dysraphism. In 30–45 % of all cases of occult spinal dysraphism it is a lumbosacral lipoma (Anderson, 1975; French, 1990; Tortori-Donati et al., 1990). If the lipomyelomeningoceles are included, then approximately every second case of spinal dysraphism can be assigned to these two disorders.
Location
Out of 100 published cases of intradural lipoma (excluding lipomyelomeningocele) the following distribution was found:
• 12 % cervical,
• 24 % cervicothoracic,
• 30 % thoracic,
• 3 % thoracolumbar,
• 3 % lumbar,
• 5 % lumbosacral,
• 2 % confined to the filum terminale.
Eight percent of tumors involved almost the entire spinal cord, 6 % the cauda equina, and 7 % the cauda equina and the conus medullaris (Giuffre, 1966).
Intradural lipomas overlie the spinal cord dorsally in about 66 % of cases, less frequently dorsolaterally (24 %), and only in exceptional cases laterally and ventrolaterally (Giuffre, 1966).
Histology
Histologically intradural lipoma is an accumulation of mature lipid cells that is, at least partially, enclosed in a capsule.
A layer of collagenous connective tissue is found at the junction between the lipoma and neural tissue.
 Lipomyelomeningocele
Lipomyelomeningocele
Definition
Lipomyelomeningoceles are complex lesions comprising spina bifida, a subcutaneous lipoma and a lumbosacral lipoma, which has contact with the dysplastic spinal cord and produces a low lying conus due to tethering of the conus. The subcutaneous lipoma is contiguous with the intradural lipoma by way of the bony defect.
At the junction between the correctly formed vertebrae and the bony defect there is always a strong fibrovascular band connecting the two vertebral arch components of the uppermost of those vertebra with a fusion defect of the arch. Those patients whose spinal canal caudal to the spina bifida is normal also demonstrate this band at the most distal of those vertebrae with a fusion defect of the arch. The band is attached to the vertebral periosteum. The meningocele and the spinal cord course dorsally below the proximal band to which the dura is sometimes firmly attached. The dorsal aspect of the spinal cord is compressed by this band and traction of the lipoma results in kinking at this point (Naidich et al., 1983).
The spinal cord itself is dysplastic, its termination flat and elongated. As with myelomeningocele, the dorsal nerve roots lie laterally to the ventral roots. The dura is attached to the placode immediately lateral to the dorsal nerve roots. The lipoma is therefore extradural and, although attached to the placode, is not invested by the dura. The arachnoid membrane courses together with the pia mater ventral to the placode. The meningocele can vary in its degree of expression and is above all found as a ventral extension of the subarachoidal space due to the dorsal displacement of the intraspinal structures. It can however also extend dorsally, lateral to the spinal cord and lipoma behind the bony defect in the spine to reach far into the subcutaneous fat.
The central canal of the spinal cord tapers off into the lipomyelomeningocele. It is open dorsally where it is sealed off by fat. The fat can extend cranially within the central canal. The lipoma is often attached to the placode posterolaterally, thus causing the spinal cord to turn to the opposite side. This rotation can displace some of the nerve roots dorsally and others ventrally, which must be kept in mind during surgery (Naidich et al., 1983).
Pathogenesis
Lipomyelomeningoceles and lumbosacral lipomas are not infrequently associated with a partial sacral agenesis (Dubowitz et al., 1965). A scimitar sacrum may be noted on radiographs. A dermal sinus is found in about 10 % of cases (Bruce and Schut, 1979).
In 30–40 % of cases telangiectases, skin appendages, increased hair growth and small skin dimples are seen in the affected region (Naidich et al., 1983).
Clinical Presentation
Not all patients show the condition clinically at the time of birth. Within their first 5 years of life about one-quarter of the patients present symptoms; some 50 % show them during the second and third decade, and 15% not until the fifth decade.
Trauma, pregnancy and physical exertion can initiate the onset of clinical symptoms (Giuffre, 1966).
Therapy
Today the generally accepted view is that the patients ought to be operated on, even if clinical symptoms are not yet apparent (Bruce and Schut, 1979; Chapman, 1982; McLone et al., 1983). The aim of surgery is to decompress the spinal cord and remove parts of the lipoma. After successful surgery these patients have a better chance of remaining free of symptoms or experiencing only mild restrictions than patients who have not been operated upon and from whom surgery is withheld until symptoms occur.
Diagnostics
MRI
MRI displays signals specific for fat on all sequences. A concomitant edematous reaction is not found, nor is there enhancement with contrast. Images in two, and preferably in all three, planes are required. Thin slices and high-resolution images should be obtained for the examination in order to detect all concomitant malformations (Figs.3.25–3.34).
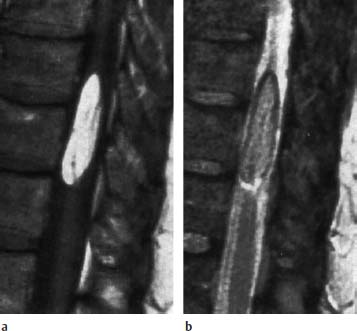
Fig. 3.25a, b Lipoma. 65-year-old patient with a thoracic lipoma overlying the spinal cord laterally. There is no tethered cord syndrome.
a T1-weighted image (SE; TR = 550 ms, TE = 20 ms): The lipoma shows a high signal intensity.
b T2-weighted sagittal section (SE; TR = 2100 ms, TE = 80 ms): Only slight signal intensity. Chemical shift artifact in the direction of the readout gradient (craniocaudal direction). There is a corresponding black, fringe-like stripe cranially and a bright stripe caudally outlining the lipoma.
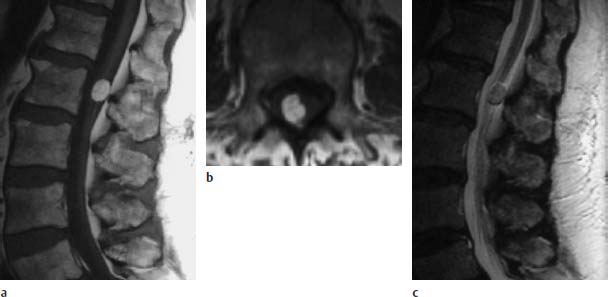
Fig. 3.26a–c Small dorsolateral lipoma at the L2 level:
a T1-weighted sagittal image (TSE; TR = 588 ms, TE = 12 ms): The rounded small lipoma is situated at the level of the conus-cauda junction. Low-lying conus is not seen.
b T1-weighted transverse image (TSE; TR = 802 ms, TE = 12 ms): The dorsolateral location of the lipoma is easily seen on this image.
c T2-weighted sagittal image (TSE; TR = 5000 ms, TE = 112 ms): The lipoma demonstrates a signal intensity similar to that of fat.
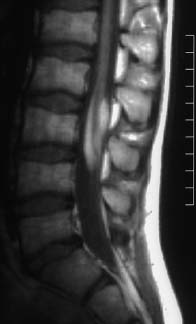
Fig. 3.27 Lumbar lipoma with concomitant lowlying conus at the L3 level. T1-weighted image (TSE; TR = 790 ms, TE = 17 ms): The lipoma envelops the spinal cord from the dorsal and ventral aspect and overlies the myelon over a long segment. It appears that there is fixation of the conus by the lipoma in the form of a tethered cord.
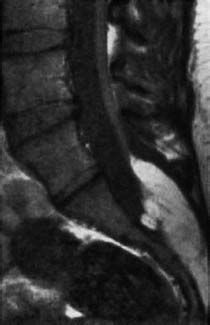
Fig. 3.28 Lipoma of the cauda equina with tethered cord syndrome. The spinal cord is elongated as far as S1. The spinal canal is markedly distended caudally (SE; TR = 680 ms, TE = 20 ms).
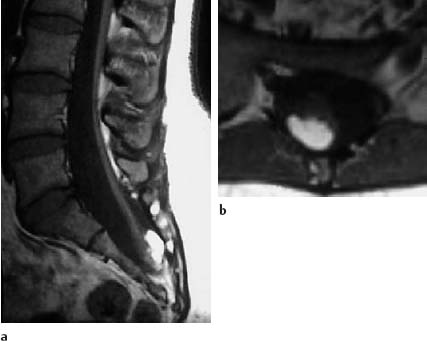
Fig. 3.29a, b Sacral lipoma with a concomitant finding corresponding to a caudal regression syndrome:
a T1-weighted sagittal section (SE; TR = 450 ms, TE = 15 ms): The spinal cord is elongated as far as S2. The lipoma overlies the conus dorsally. The sacrum is markedly shortened in a caudal direction. The sacral canal appears enlarged.
b Transverse section (SE; TR = 580 ms, TE = 15 ms): The dorsal location of the lipoma is shown well in this slice orientation.
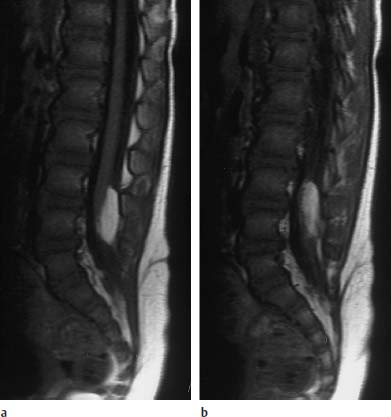
Fig. 3.30a, b Intradural lipoma at the level of L4 with associated malformation (dermal sinus):
a T1-weighted sagittal section (TSE; TR = 800 ms, TE = 12 ms): The lipoma overlies the spinal cord dorsally at the level of L4. Note the concomitant dermal sinus traversing in a dorsal direction.
b Slice 3mm parallel to a: The further course of the dermal sinus is well noted on this image. The sinus ends at the level of the bone, an intraspinal extension was not detectable, neither in the conventional images shown nor in the (not depicted) contrast-assisted images.
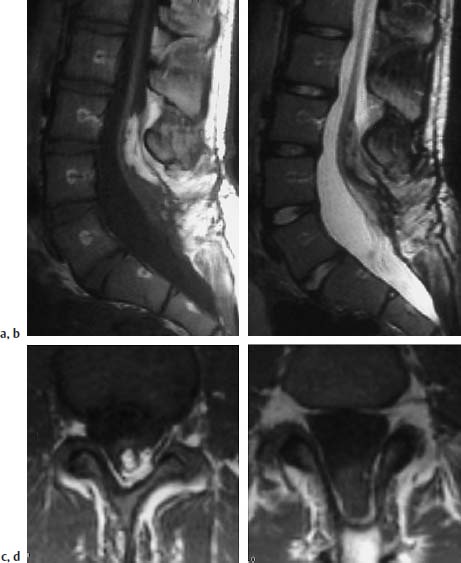
Fig. 3.31a–d Lipomyelomeningocele:
a T1-weighted sagittal section with evidence of an intradural lipomatous component at the level L3/L4. This lipoma communicates with the extradural part. Note the meningocele-like enlargement of the spinal canal, particularly dorsally. The spinal cord extends distally as far as L5/S1 where it exhibits a dorsal attachment to the dura.
b Corresponding T2-weighted image (TSE; TR = 5000 ms, TE = 112 ms): The lipoma is poorly delineated from the spinal cord components on this image. The nerve roots coursing ventrally at the level of L5/S1 can be seen easily.
c T1-weighted transverse image (SE; TR = 580 ms, TE = 15 ms): Dorsal lipoma with tethering of the spinal cord.
d Image 2 cm distal to c: Spina bifida with dorsal opening of the spinal canal. Meningocele-like enlargement. The spinal cord can be seen as a coarse structure in the dorsal segments of the spinal canal with attachment to the dura. Note the fringe-like epidural fat in the dorsal parts of the meningocele.
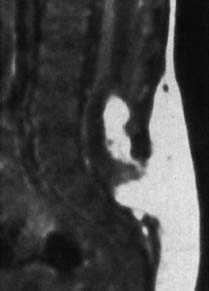
Fig. 3.32 Lypomyelocele. 3-week-old boy. Subcutaneous and intradural lipomas communicating with each other via the bony defect. Tethering of the spinal cord and low-lying conus at the level L4. No meningocele (SE; TR = 650 ms, TE = 25 ms).
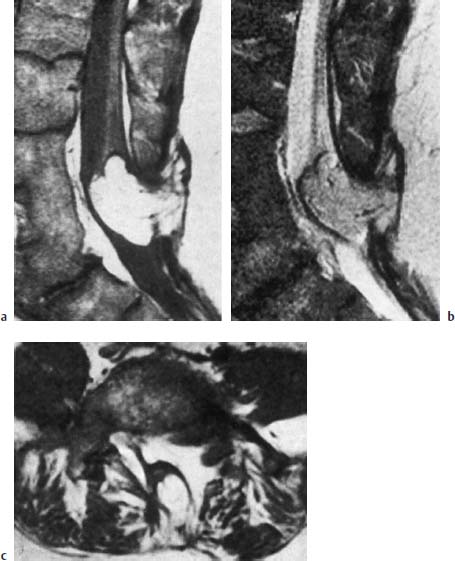
Fig. 3.33a–c Lipomyelomeningocele. 41-year-old patient:
a Malformation of the distal three lumbar vertebrae with evidence of vertebral shortening and a considerable degree of fusion. Extensive subcutaneous lipoma communicating with the intradural component. Tethering of the low-lying conus. Enlargement of the spinal canal and evidence of a corresponding spinal bifida at the L4–L5 level (SE; TR = 750 ms, TE = 25 ms).
b T2-weighted image (SE; TR = 2100 ms, TE = 80 ms).
c Transverse section at the L5 level (SE; TR = 550 ms, TE = 25 ms).
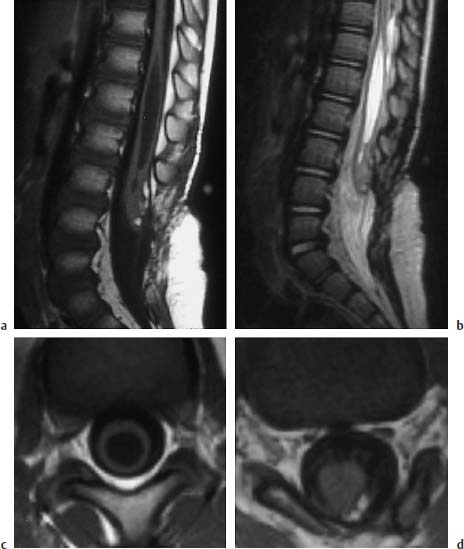
Fig. 3.34a–d Postoperative lipomyelomeningocele at the level of L3:
a T1-weighted sagittal image (TSE; TR = 600 ms, TE = 12 ms): Extensive spina bifida reaching from L3 to S1. The caudal part of the spinal cord appears thickened at the level of L3. A small lipomatous remnant can be seen dorsally. The spinal cord appears dorsally attached to the dura. Large syrinx, in part with a distinct distension of the myelon.
b T2-weighted sagittal image (TSE; TR = 5000 ms, TE = 112 ms): The extent of the syrinx is easily identifiable due to its high CSF signal. The filum is seen as a thin linear structure, although it is evidently not stretched out.
c T1-weighted transverse section with T1-weighting at the level of the syrinx (SE; TR = 580 ms, TE = 15 ms): Marked ballooning of the spinal cord with corresponding pressure atrophy.
d Section at the level of a small lipomatous remnant (SE; TR = 580 ms, TE = 15 ms): The lipoma overlies the spinal cord with a fibrous component dorsally and extends laterally to the left. Note the nerve roots on either side evidently exiting a typically and dorsally from the spinal cord and also appearing slightly thickened.
 Fibrolipomatosis of the Filum Terminale
Fibrolipomatosis of the Filum Terminale
The filum is not infrequently divided into a second cord, which runs dorsally into the skin, giving rise to a small, circumscribed dimple (Emery and Lendon, 1969).
The filum is normally as much as 2mm thick in diameter (measured at the level L5/S1).
Fibrolipomatosis of the filum terminale can develop from faulty regression at a lumbosacral level during the embryonic phase. This becomes evident from a thickening of the filum and can lead to shortening and consequently to a lowlying conus as a result of conus tethering. Occasionally however, the conus is found at a normal position despite the formation of fibrolipomatosis of the filum terminale. Fibrolipomatosis is found both in exclusively extradural and intradural-extradural locations, as well as exclusively extradurally.
The lipoma has a sausage-like configuration with intradural-extradural expansion and tapers at the site of penetration through the dura. The extradural component is not infrequently larger than the intradural part and can demonstrate a considerable expansion with infiltration of the surrounding tissue when occurring exclusively extradurally. The intradural lipoma normally has a capsule (Emery and Lendon, 1969).
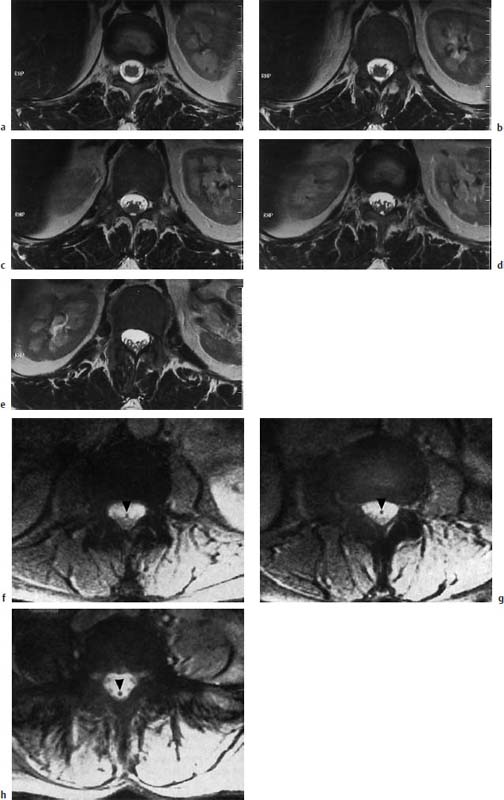
Fig. 3.35a–h Normal conus-cauda junction. Transverse sections (TSE; TR = 6310 ms, TE = 120 ms):
a–e The transverse images show the nerve roots exiting from the conus and the junction to the cauda. The segmental spidery configuration is easily seen.
h Depiction of the filum (arrowhead) which stands out well as a dark spot due to discrete fibrolipomatosis (GRE; TR = 680 ms, TE = 20 ms, flip angle α = 10°). No elongation of the conus. Due to the lack of lipomatosis the filum is demarcated neither on T2-weighted GRE images nor on T1-weighted images.
Diagnostics
MRI
The conus-cauda junction is well defined on T2-weighted transverse images. On T2- and T1-weighted images the filum is normally indistinguishable in its signal from the nerve roots, but delineation is clearly possible in the presence of fibrolipomatous transformation (Fig. 3.35).
Fibrolipomatosis is easily recognized on T1-weighted images by its characteristic fat signal. Here sagittal and transverse images should be obtained to show its entire course.
For depiction of fibrolipomatosis, T2-weighted images should be taken using a GRE technique because the lipomatous components are well delineated from the cauda due to the complete loss of signal. The differentiation between cauda and fibrolipomatosis is often impossible using T2-weighted SE sequences (Figs. 3.36–3.38).
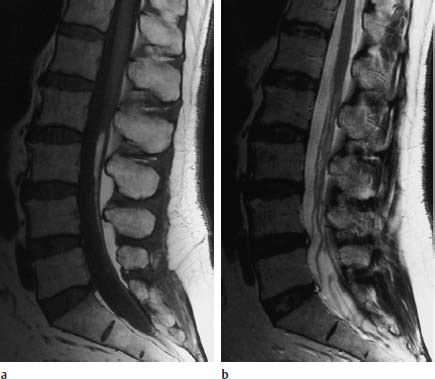
Fig. 3.36a, b Fibrolipomatosis with an obvious enlargement of the filum terminale at the level L3.
a T1-weighted sagittal image (TSE; TR = 800 ms, TE = 12 ms): Well-defined filum with corresponding fibrolipomatous modification with a distinct thickening, recognizable particularly at the level L3. The caudal parts of the filum also appear somewhat thickened, albeit without deposits of fatty tissue. The conus is located at the level L1/L2 and is therefore not lowlying.
b Corresponding T2-weighted image using SE technique (TSE; TR = 4000 ms, TE = 120 ms): On this image the lipomatous modification is not recognizable.
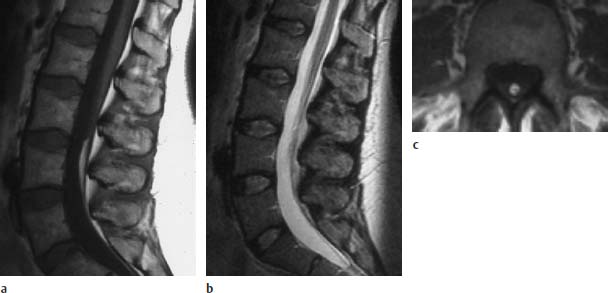
Fig. 3.37a–c Localized fibrolipomatosis at the level L4:
a T1-weighted sagittal section (TSE; TR = 588 ms, TE = 12 ms): Fibrolipomatous modification of the filum with typical fat signal.
b Corresponding T2-weighted image using an SE technique (TSE; TR = 5000 ms, TE = 112 ms): The fibrolipomatosis is not clearly recognizable on this image.
c T1-weighted transverse section (SE; TR = 580 ms, TE = 15 ms): Well-defined fibrolipomatous structure.
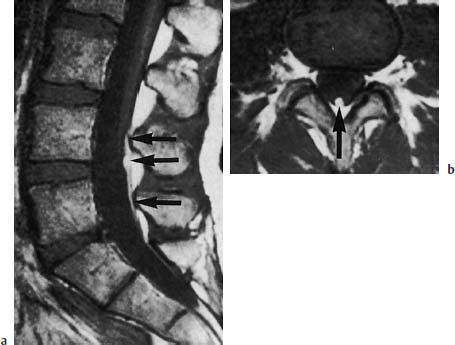
Fig. 3.38a, b Fibrolipomatosis with tethered cord syndrome. 37-year-old patient:
a The sagittal section shows evidence of tapering of the conus with low-lying spinal cord at the level L4 (arrow) (SE; TR = 550 ms, TE = 20 ms).
b The transverse section also shows the lipomatous modification and thickening of the filum to a diameter of 4mm (arrow) (SE; TR = 550 ms, TE = 20 ms).
 Tethered Cord Syndrome
Tethered Cord Syndrome
Definition and Pathogenesis
In a broad sense, this term embraces all disorders that are associated with lumbosacral tethering of the spinal cord and result in a low-lying conus.
In these disorders, therefore, the conus does not reach its normal position at the level of L1/L2 that is found in healthy individuals about 8–10 weeks after birth.
The spinal cord, and especially the conus medullaris, demonstrates a change in form in these cases. The conus appears thin and elongated; the normal thickening of the spinal cord at the level of the conus is not found.
The tethered cord syndrome is caused by all disorders that result in this tethering of the conus:
• lumbosacral lipoma,
• lipomyelomeningocele,
• fibrolipomatosis of the filum terminale,
• split cord malformation,
• dermal sinus,
• intradural congenital tumors.
In a narrower sense, the tethered cord syndrome is a disorder brought about by an alteration of the filum terminale.
The filum is usually about 24 cm long and up to 2mm thick. The tethered cord syndrome involves a shortening and thickening of the filum, which results in tethering of the conus in an abnormally low position distal to L2. The thickening of the filum is frequently associated with the formation of bands connecting the cauda equina and dura together in the lumbosacral space.
The clinical picture is very rare and is seen in only 2 % of all patients with occult spinal dysraphism in large studies (Jones and Love, 1956).
Clinical Presentation
The patients often present with skin alterations as well as increased hair growth or a subcutaneous lipoma.
Bony alterations are not infrequently encountered, including:
• disturbances of vertebral fusion,
• enlargement of the spinal canal,
• scoliosis,
• malformation of the ribs,
• block vertebrae.
The patients develop clinical manifestations during childhood, rarely at the time of birth.
Therapy
The therapy of choice is the removal of the bandlike adhesions and division of the shortened filum terminale (Jones and Love, 1956).
Diagnostics
MRI
Sagittal and transverse T1-weighted images demonstrate the thickening of the filum, displaying a signal typical for fibrous tissue. Lipomatous components are lacking. The spinal cord appears elongated, the conus stretched (Figs. 3.39–3.41).
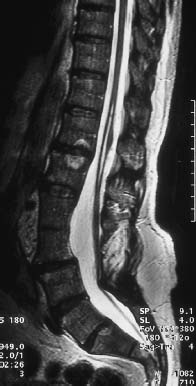
Fig. 3.39 Thickened and shortened filum terminale with low-lying conus and corresponding thinning and elongation of the conus. In addition, there is evidence of a syrinx in the region T12-L2 and incompletely fused vertebrae in the region L3/4 (TSE; TR = 3950 ms, TE = 112 ms).
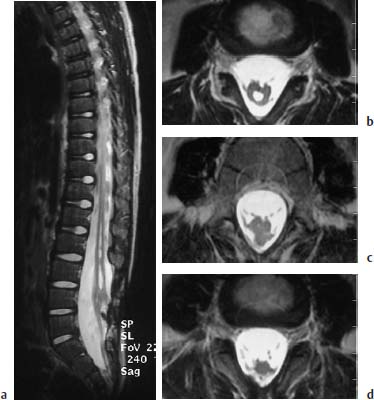
Fig. 3.40a–d Tethered cord syndrome. Tethered cord syndrome in the presence of a complex malformation with evidence of spina bifida L3–S1, marked shortening and thickening of the conus at the L4 level, a syrinx in the region L1–L3 and an enlargement of the spinal canal in the region L3–S1:
a
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


