Fig. 8.1
NUT midline carcinoma. Histology consists of an undifferentiated neoplasm with vague epithelioid features
Prognostic Features
With few exceptions, NUT carcinomas are highly lethal malignancies. Most patients die within weeks, as metastases are typically present at diagnosis, and bulky intrathoracic growth produces superior vena cava compression. No association of outcome with fusion type has been demonstrated to date [22].
Squamous Cell Carcinoma (SCC)
HPV-related squamous cell carcinomas are discussed in the head and neck chapter (Chap. 7).
Definition
Type of bronchogenic malignancy of the lung.
Clinical Features and Epidemiology
Squamous cell carcinoma (SCC) is a common malignancy in adults, accounting for 44 and 25 % of primary malignancies of the lung of men and women, respectively [26]. In adults there is a strong association with smoking.
Bronchogenic carcinoma is much less common in children than in adults, accounting for 7–17 % of primary pediatric pulmonary malignancies [4, 5]. When seen in children, SCC is often associated with recurrent respiratory papillomatosis (RRP).
RRP is caused by the human papillomavirus (HPV) transmitted primarily via vaginal delivery [27]. RRP is usually isolated to the larynx (>95 % of cases). The distal trachea is affected in less than 5 % of cases and the pulmonary parenchyma is affected in less than 1 % of cases [28]. While pulmonary involvement is uncommon, papillomas are the most common primary benign tumor of the lower respiratory tract in children (accounting for up to 40 %) [5]. Distal dissemination may be caused by aerogenous embolization of papilloma particles and can be precipitated by tracheotomy or other airway procedures [29]. Juvenile-onset RRP has a bimodal peak at 2 and 10 years of age [30]. No clear sex predilection has been identified.
RRP undergoes malignant transformation into SCC in less than 1 % of cases [28]. Transformation can occur with or without a history of smoking or radiation, and is most frequently observed in males in the third and fourth decades of life.
Imaging Features
RRP can present as a single or, more commonly, multiple masses predominantly affecting the airway and posterior lower lobes [29]. The masses may be solid or cavitary with a thin or thick wall and either air or fluid contents (Fig. 8.2). An air-fluid level is a nonspecific finding that may be seen in the setting of superimposed infection or hemorrhage. Benign lesions may demonstrate increased FDG uptake on PET imaging and simulate malignancy [30]. Findings that suggest malignant transformation include lesion growth, thoracic lymphadenopathy, or distal metastases.
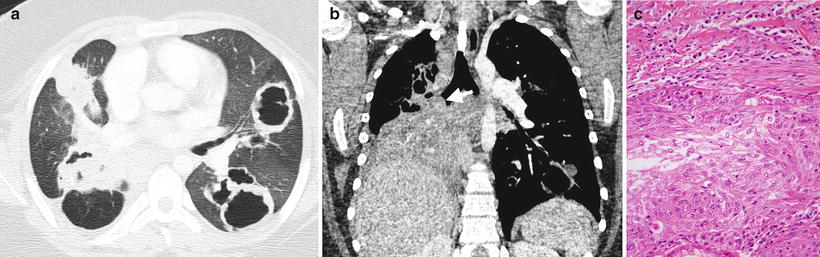
Fig. 8.2
Recurrent respiratory papillomatosis. Axial (a) and coronal (b) contrast-enhanced chest CT images from a 13-year-old male demonstrate multiple thick-walled cavitary lung lesions and an endobronchial papilloma (arrow) occluding the bronchus intermedius with post-obstructive atelectasis of the right middle and lower lobes. Histology (c) of another case shows a well-differentiated squamous carcinoma invading the underlying tissues of the bronchial submucosa and inciting adjacent fibrosis
Associated findings include atelectasis, consolidation, and bronchiectasis secondary to airway obstruction and recurrent superimposed infections [29].
Molecular Genetics
Respiratory papillomatosis is usually caused by low-risk HPV types 6 and 11. Certain HPV types, particularly types 16 and 18, are more likely to transform into carcinoma, similar to uterine cervical cancers. With HPV16-associated lesions, transformation is related to genetic polymorphisms or deletions, but aggressiveness of low-risk HPV11-associated lesions does not relate to intratypic variants but rather to the interaction of multiple genetic factors [31].
Pathology
The pathological features of HPV-related carcinomas are discussed in the head and neck chapter (Chap. 7) (Fig. 8.2). Juvenile papillomas from the upper respiratory tract may be aspirated into the bronchial passages. From there, they may act similar to metastatic lesions, as malignant transformation may produce a well-differentiated squamous carcinoma.
Prognostic Features
RRP associated with HPV 11 demonstrates a more aggressive course and worse prognosis [32].
Endobronchial Tumors
Bronchial Carcinoid
Definition
Malignant neuroendocrine tumor of endodermal origin arising from Kulchitsky cells.
Clinical Features and Epidemiology
Neuroendocrine and salivary gland tumors of the tracheobronchial tree were previously referred to as bronchial adenomas [1]. This potentially confusing terminology has fallen out of favor secondary to the variable glandular component and malignant potential of these tumors [33].
Bronchial carcinoid is the most common malignant endobronchial tumor and the second most common primary malignancy of the lower respiratory tract in children (after PPB) [5]. Carcinoids are most commonly seen in the gastrointestinal tract, with only 10–32 % occurring in the tracheobronchial tree [34, 35]. The average age of presentation of bronchial carcinoid is 45–56 years [34, 35]. This is a decade younger than the average age of presentation for other lung malignancies, but carcinoid is still uncommon in the pediatric age range. When it occurs in children, it tends to affect older children and adolescents with the youngest reported patient being 8 years of age [34]. There is no known smoking association, or sex or racial predilection.
Presentation is varied. Post-obstructive atelectasis and pneumonitis are common [35]. Nonspecific respiratory symptoms and signs, including pleuritic pain, dyspnea, cough, wheeze, and hemoptysis, are also seen. The tumor is incidentally detected in up to half of patients. Carcinoid tumors can synthesize a variety of neuroamines and peptides leading to more specific presentations. Overproduction of adrenocorticotropic hormone (ACTH) leads to Cushing syndrome in up to 4 % of patients. Overproduction of serotonin leading to carcinoid syndrome is rare in bronchial carcinoid outside of the setting of metastatic disease (2–5 %). Up to 4 % of carcinoid tumors are associated with other endocrine neoplasias [34]. The most common association is with pituitary tumors, seen in half of this patient subset.
In addition to imaging, detection and follow-up of lesions can be aided by serum and urine 5-hydroxyindolacetic acid (5-HIAA), a serotonin metabolite that is relatively specific though not sensitive for the diagnosis [34].
Imaging Features
The typical appearance of a bronchial carcinoid is that of a single round or lobulated mass that is at least partially endobronchial [35, 36]. The mass is typically 2–5 cm in size. Endoscopy can underestimate the size of the mass as the endobronchial component can represent just the “tip of the iceberg.” The mass is seen within the main, lobar, and segmental bronchi in 80 % of cases with a preference for branching sites (Fig. 8.3). The remaining 20 % of bronchial carcinoids are peripheral in location and indistinguishable from intraparenchymal pulmonary masses [35].
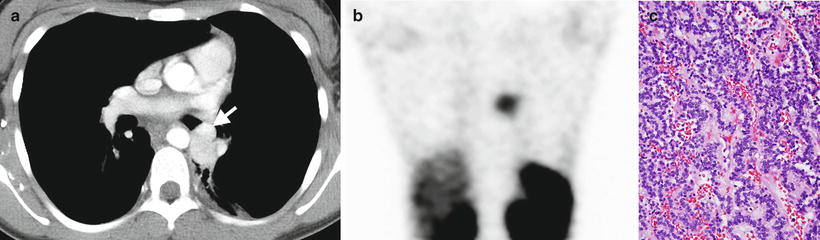
Fig. 8.3
Bronchial carcinoid. An axial contrast-enhanced chest CT image (a) from a 13-year-old female shows a hyper-enhancing lobular soft-tissue mass with a “tip of the iceberg” endobronchial component (arrow) involving the distal left mainstem bronchus. A coronal image (b) from an In-111 octreotide SPECT scan shows radiopharmaceutical uptake by the mass. Histology (c) of another case demonstrates uniform cells with round nuclei, forming cords and festoons
In up to 30 % of cases, bronchial carcinoids demonstrate calcification that is best appreciated on CT. The masses demonstrate high signal intensity on T2-weighted MRI images and typically exhibit homogeneous avid enhancement [35]. However, heterogeneous or minimal enhancement does not exclude the entity. Bronchial carcinoids demonstrate octreotide uptake on somatostatin receptor scintingraphy in greater than 86 % of cases [35, 37] (Fig. 8.3).
Associated findings include post-obstructive atelectasis or pneumonitis, bronchiectasis, and mucous plugging. Local or distal recurrence occurs in up to a fifth of cases with 15 % presenting with metastases [35]. Metastases are most commonly seen in the liver, bone, adrenals, and brain.
Molecular Genetics
In the current molecular model [38], bronchial carcinoids originate from bronchial Kulchitsky-type neuroendocrine cells. These cells are capable of producing precursor lesions such as diffuse idiopathic pulmonary cell hyperplasia or tumorlets. Small invasive foci break through the basal lamina and invade locally, producing small lesions termed tumorlets, which are by definition <5 mm in diameter. The most common chromosomal aberrations in pulmonary carcinoids include −11p, +19p, −13q, +19q, +17q. −11p, −6q, +16p, +2-p, and −3p, although these occur at a frequency of <25 % in typical lesions. However, as bronchial carcinoids assume atypical features and undergo carcinomatous transformation, the frequency of these changes rises to as high as 75 % for −3p and −13q. Approximately 18 % show sporadic mutations and loss of heterozygosity of MEN1, the multiple endocrine neoplasia 1 gene.
Pathology
Pulmonary carcinoids generally contain a uniform population of monotonous round cells with regular, central nuclei, granular chromatin, inconspicuous nuclei, and a relatively high nuclear-to-cytoplasmic ratio. The cytoplasm is generally lightly eosinophilic but on occasion is more abundant and resembles that of rhabdoid cells. Rarely, it is clear, and melanin or mucus may be present. Some tumors show marked nuclear pleomorphism, a feature not reliable in predicting behavior. The cells usually grow in an organoid or trabecular pattern, forming nests or cords, but sometimes they form spindle cells or rosettes. The intervening stroma is vascular and collagenous and is capable of producing amyloid, hyaline, bone, or cartilage (Fig. 8.3).
Atypical carcinoids are characterized by the presence of focal necrosis or conspicuous mitotic activity (2–10 mitoses/2 mm2) [33].
Prognostic Features
Prognostic factors in bronchial carcinoids include stage and the presence of atypical features (atypical carcinoid). Pediatric bronchial carcinoids generally have an excellent long-term outcome with adequate surgery and lymph node excision. Long-term follow-up is required, as relapse may occur years after the initial excision, but usually can be successfully treated with additional surgery [39].
Mucoepidermoid Carcinoma
Definition
Malignant salivary-type neoplasm arising from the mucous cells of the submucosa.
Clinical Features and Epidemiology
Mucoepidermoid carcinoma comprises 0.1–0.2 % of malignant lung tumors and 2.5–7.3 % of endobronchial tumors [34]. While it is even more unusual in children, it is likely the second most common endobronchial malignancy [34]. The tumor has a wide age range of 3–78 years, with half of cases occurring in patients younger than 30. The tumor may be more common in Caucasians and has a 3:2 male-to-female ratio [33].
Obstructive airway symptoms and signs are a common presentation and include wheezing and recurrent pneumonia. Up to 25 % of patients are asymptomatic and the tumor is incidentally discovered [34].
Mucoepidermoid carcinoma is graded by histology similar to other salivary-type neoplasms into low, intermediate, and high grades. Higher grades have a higher rate of metastasis and worse prognosis [40].
Imaging Features
The typical appearance is a single round, lobulated or polypoid mass occurring in a proximal lobar bronchus [41] (Fig. 8.4). A more peripheral mass with the same histology should raise concern for metastatic salivary gland neoplasm [34]. The mass is typically 1–4 cm in size and often extends beyond the lumen of the bronchus. Calcification is seen in up to half of cases. Grades are indistinguishable by imaging.
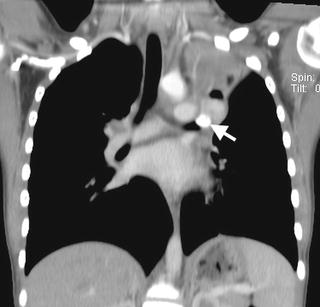
Fig. 8.4
Mucoepidermoid carcinoma. A coronal contrast-enhanced chest CT image from a 12-year-old male depicts a partially calcified endobronchial mass (arrow) occluding the left upper lobe bronchus with associated left upper lobe collapse
These endobronchial tumors frequently cause airway obstruction with associated findings of atelectasis, bronchiectasis, and mucous plugging. Post-obstructive pneumonitis is seen in up to a third of cases [42].
Lymph node extension and distal metastasis are infrequent, being seen in less than 10 % of cases [42].
Molecular Genetics
The molecular genetic features of mucoepidermoid carcinoma are discussed in the head and neck chapter.
Pathology
The pathological features of mucoepidermoid carcinoma are discussed in the head and neck chapter.
Prognostic Features
Pediatric mucoepidermoid carcinomas of the lung are generally low-grade, low-stage lesions with excellent patient survival following excision. In a recent SEER survey [43], all 14 patients survived. In a recent Egyptian study of radiographic features [44], overall survival of patients with salivary gland-type tumors of the lung was affected by the presence of mediastinal/hilar lymphadenopathy, suspected metastatic disease, and primary tumor heterogeneity. Higher FDG uptake on PET was associated with nodal metastasis. The mean survival time for pulmonary mucoepidermoid carcinoma was 4.4 years. In a recent Chinese study [45], prognostic factors for pulmonary mucoepidermoid carcinoma included age, grade, lymph node metastasis, and stage. Lower age had a positive effect on outcome, with 80 % survival in patients <60 years old.
Mesenchymal Tumors
Pleuropulmonary Blastoma
Definition
Pleuropulmonary blastoma (PPB) is a rare primitive mesenchymal, embryonal type neoplasm of the lung and pleura of young children.
Clinical Features and Epidemiology
PPB has been previously termed pulmonary embryoma, pulmonary blastoma, mesenchymal cystic hamartoma, and sarcoma arising in congenital cystic malformations [5]. PPB was described as a tumor distinct from adult pulmonary blastoma in 1988 by Manivel et al. [46]. PPB is a rare neoplasm with approximately 25–50 cases occurring annually in the USA, 500–600 cases reported in the literature, and 370 centrally reviewed cases in the International Pleuropulmonary Registry (Personal communication, Dr. Yoav Messinger, on July 17, 2013). In spite of its rarity, it is likely the most common primary malignancy of the lung in children. Epidemiological data is largely derived from the largest reported case series of 50 patients from the Registry [47]. The average age of presentation is 38 months with the cystic types presenting earlier and rarely presenting at more than 6 years of age. Prenatal presentation of cystic PPB has been reported [48].
There is no apparent sex or racial predilection. About 40 % of patients with PPB or their relatives manifest certain other dysplasias and neoplasms as part of the PPB Family Tumor Dysplasia Syndrome (PPB-FTDS). Conditions associated with PPB-FTDS include PPB, cystic nephroma, pineoblastoma, pituitary blastoma, embryonal rhabdomyosarcoma (especially of the uterine cervix), medulloblastoma, nasal chondromesenchymal hamartoma, ciliary body medulloepithelioma, multinodular goiter, intestinal juvenile hamartomatous polyp, and ovarian stromal sex-cord tumor (especially Sertoli-Leydig tumors) [49–51]. The clinical presentation of PPB ranges from an incidental finding in an asymptomatic patient to nonspecific respiratory or systemic symptoms, such as respiratory distress, fever, chest pain, cough, anorexia, and malaise [47].
Imaging Features
PPB has been categorized into three main types based on gross morphology. Type I is entirely cystic, type II is mixed cystic and solid, and type III is entirely solid [52]. Age of presentation, malignant behavior, and mortality increase with type number. If untreated, progression of type I to type II and III can occur. CPAMs do not transform into PPB [47]. Type I PPB may also spontaneously regress, leading to categorization as type Ir PPB for type I-regressed PPB [49].
Types I and Ir PPB appear as an air-filled unilocular or multilocular thin-walled cyst with delicate septations, and are indistinguishable from the more common large cyst form of CPAM (Fig. 8.5). Findings that favor type I PPB over large-cyst CPAM include the presence of multifocal or bilateral cysts or spontaneous pneumothorax [47–49, 53]. Type II and III PPB usually appear as a large, heterogeneously enhancing mass in the hemithorax with associated mediastinal mass effect and pleural effusion and lack of chest wall invasion [9] (Fig. 8.6). PPB is slightly more common in the right hemithorax than the left, and bilateral tumors may be synchronous or metachronous [54].
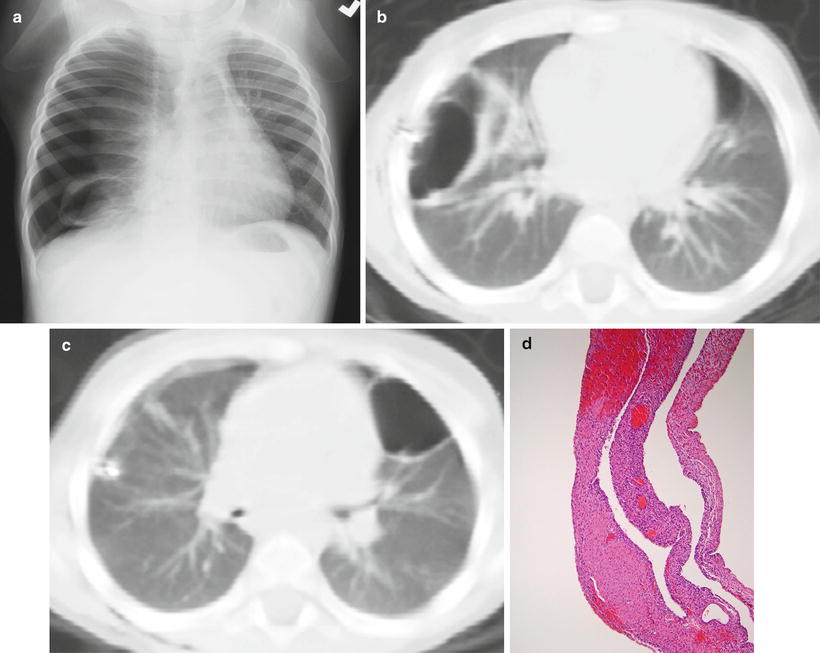
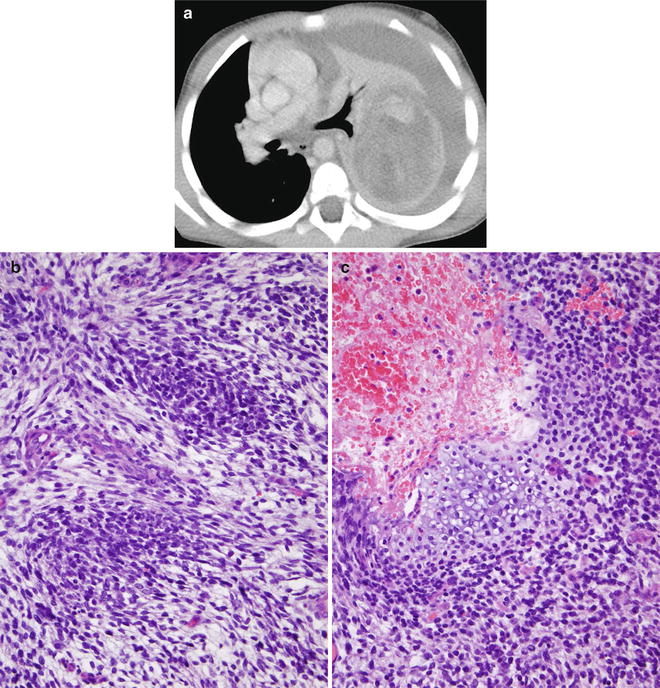
Fig. 8.5
Type I (cystic) pleuropulmonary blastoma. A chest radiograph (a) from a 2-year-old presenting with respiratory distress demonstrates a right pneumothorax, right lung collapse, and a cystic lesion of the right lower lung. Axial chest CT images (b, c) obtained after right chest tube placement and pneumothorax evacuation demonstrate bilateral air-filled, thin-walled cystic lung lesions. Histology (d) of another case shows large spaces lined by thin septa, some containing nodules of primitive blastemal cells
Fig. 8.6
Type III (solid) pleuropulmonary blastoma. An axial contrast-enhanced chest CT image (a) from a 3-year-old male reveals a large, heterogeneously enhancing left pulmonary mass with a large left pleural effusion and rightward mediastinal shift from mass effect. Histology (b) of another case demonstrates primitive blastemal cells arranged in an organoid pattern. A small bar of hyaloid cartilage is revealed in an additional histologic section (c)
Aggressive local invasion of the bronchi, great vessel, and heart is unusual but has been reported [54, 55]. Metastases occur in 11 % and 55 %, respectively, of types II and III cases [49]. The most common location for distal metastasis is the brain, followed by bone. Cerebral metastases are much more frequent in PPB than in other childhood sarcomas [56]. Metastases to the liver, adrenal glands, and ovary have also been reported [47, 49].
Molecular Genetics
Comparative genomic hybridization identifies aberrations (amplifications, gains, losses) in PPB tumors, with DNA gains involving chromosome 8q being the most frequent abnormality. Losses of 9p and 11q are also reported [57]. The sites of gains and losses may contain oncogenes or tumor suppressor genes, respectively.
Children with PPB-FTDS discussed above frequently have heterozygous, germline loss-of-function mutations in the DICER1 gene that codes for a protein involved in microRNA (miRNA) processing. Loss of DICER 1 function in the epithelium of the developing lung may alter the regulation of diffusible factors that results in mesenchymal proliferation and sarcomatous transformation [58]. DICER1 mutations are inherited by an autosomal dominant mechanism with variable expressivity, and are thought to be present in 60–70 % of cases of PPB [49].
Pathology
PPB is composed of a malignant mesenchymal component but no malignant epithelial component. It is postulated that PPB originates in mesenchymal elements resembling fetal lung at 10–16 weeks of gestation. Three types (I, II, and III) based on gross morphology are described [59].
A unilocular or multilocular cyst with thin fibrous septa characterizes type I PPB. The cysts are lined by ciliated epithelium with small subepithelial “buds” containing aggregates of primitive mesenchymal cells or nodules of immature cartilage (Fig. 8.5). These small, round-to-spindled, subepithelial mesenchymal cells may display rhabdomyoblastic differentiation. In type Ir (regressed cystic), the small primitive mesenchymal cells are not seen, and the wall or septa of the cyst may be hyalinized or necrotic.
In type II PPB, cystic and plaque-like areas mingle with solid areas with overgrowth of rhabdomyoblasts, spindle cell sarcoma, or blastematous elements. Type III PPB consists of a solid tumor with mixed sarcomatous (fibrosarcoma, rhabdomyosarcoma, pleomorphic undifferentiated sarcoma) and blastematous features (Fig. 8.6). Foci of muscle and chondroid differentiation are frequent, as are foci of anaplasia with giant bizarre pleomorphic tumor cells (Fig. 8.6a). Malignant epithelium is not seen, differentiating PPB from pulmonary blastoma. Immunohistochemical staining is variable from one tissue type to another. Myogenin, desmin, and MyoD1 are useful for the identification of rhabdomyoblasts [49].
As was mentioned, PPB is histologically and clinically distinct from pulmonary blastoma, a rare subtype of malignant biphasic sarcomatoid carcinoma typically seen in adults. However, a recent case report of a neonate described histology distinct from PPB and more similar to adult PB [60]. PB is occasionally reported in adolescents [61].
Prognostic Features
Overall survival rate is estimated at 90 % for type I and 40–60 % for types II and III [49]. Progression of type I to type II and III is well recognized. Tumors do not regress from type III to type II or from type II to type I. Local recurrence develops in fewer than 15 % of type I but is seen in over 45 % of types II and III [49]. Recurrence can affect the ipsilateral or contralateral lung [47]. Sex, tumor side, tumor size, preexisting lung cysts, and extent of surgical resection at the time of diagnosis do not impact prognosis, whereas incomplete resection and extrapulmonary involvement at diagnosis result in a significantly worse prognosis [62].
Fetal Interstitial Lung Tumor (FLIT)
Definition
Tumefactive lesion presenting in utero or early infancy with a solid appearance on imaging and gross inspection and a prominent immature interstitium on histology that resembles immature fetal lung at 20–24 weeks gestational age.
Clinical Features and Epidemiology
FLIT is a rare tumor recently described in a series of ten cases [63]. All were discovered before 3 months of age. Two were detected by prenatal ultrasound and one of these was further evaluated by prenatal MRI [64]. In the patient that underwent MRI, the tumor was not seen at routine second trimester prenatal ultrasound but was first noted in late third trimester as a large pulmonary mass causing hydrops, prompting ex utero intrapartum treatment (EXIT procedure) at 37 weeks gestational age. This late gestational growth and late onset of hydrops would be atypical for a CPAM or PPB [64]. The other patients presented with respiratory symptoms, two in the immediate postpartum period [63].
Imaging Features
The tumors are well circumscribed, unifocal, and confined within a single lobe. The tumors have been reported in all lobes, with the right lower lobe being the most common location [63]. The tumors usually appear solid, with small cysts occasionally observed. On prenatal ultrasound, the tumors are hyperechoic compared to normal lung parenchyma (Fig. 8.7). The tumors are predominantly low in attenuation on CT. The single case examined by MRI appeared heterogeneously hyperintense on T2-weighted images, with mass effect compressing the heart and inferior vena cava leading to fetal hydrops (Fig. 8.7). The solid nature of a FLIT can resemble a type 3 CPAM, sequestration, or congenital peribronchial myofibroblastic tumor.
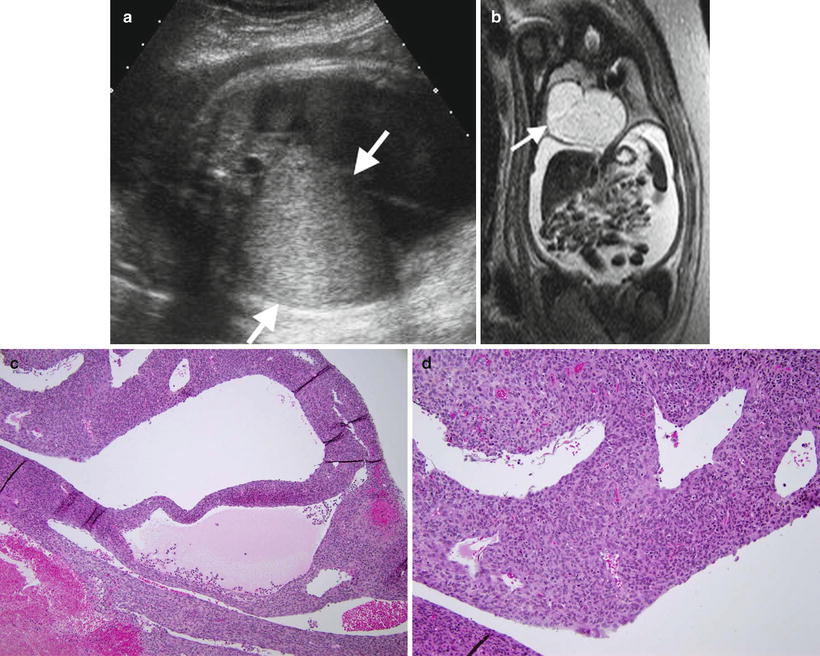
Fig. 8.7
Fetal lung interstitial tumor. Prenatal ultrasound (a) at 36 weeks gestation shows a large, well-circumscribed, solid hyperechoic mass arising from the right lung (arrows). T2-weighted fetal MRI image (b) at 36 weeks gestation shows the high signal intensity mass compressing the adjacent lung and everting the diaphragm, with associated hydrops and large-volume ascites from inferior vena cava compression. Histology (c) of another case shows primitive mesenchymal cells that line spaces resembling alveoli. An additional histologic section (d) demonstrates constituent cells that are primitive and lack obvious differentiation
Although rapid growth can be observed, no locally recurrent or metastatic FLIT has been described to date, even in the setting of an incompletely resected lesion [63].
Molecular Genetics
No genetic aberrations have been reported. The one child in the defining series who underwent testing for DICER1 mutations associated with PPB tested negative [63].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


