Fig. 4.1
Plain film radiograph of conventional high grade osteosarcoma of the femur in a 14 year old girl. Note the osteoid matrix of the large soft tissue mass and Codman’s triangle (arrow)
Computed tomography (CT): CT is superior to plain film radiography for showing marrow involvement and defining osteoid matrix, which can be difficult to detect on MRI in predominantly lytic lesions. CT is rarely necessary in the evaluation of the primary tumor but aids in staging of pulmonary metastases and guiding minimally invasive biopsies (CT-guided biopsy). It is superior to plain film radiography at depicting the low attenuation of central necrosis in soft tissues.
Magnetic resonance imaging (MRI): MRI is useful for soft tissue and marrow evaluation, surgical or biopsy planning, and restaging after chemotherapy (Fig. 4.2a–c). Osteoid appears as a low signal on all sequences, whereas non-osteoid, solid portions of the tumor appear isointense to skeletal muscle on T1W sequences. Fluid-sensitive sequences show heterogeneous high-signal features in both the soft tissue and bony masses [15]. High-signal peritumoral edema in bone and soft tissue may exaggerate the size of the mass. Post-contrast images reveal intense, heterogenous enhancement of the soft tissue and involved marrow, which may help differentiate viable regions from low-intensity necrotic regions.
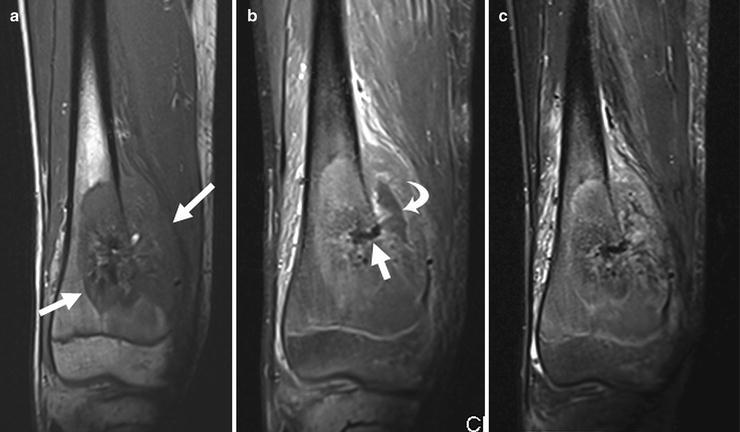
Fig. 4.2
MRI images of same tumor shown in Fig. 4.1. (a) Coronal T1W fat-saturated image: the intramedullary extent of the tumor is well delineated. The lesion is hypointense to marrow (arrows). The center of the tumor is dark due to dense calcification. (b) Post-contrast T1W fat-saturated image demonstrating enhancement of the intramedullary tumor and soft-tissue component. The central calcification (straight arrow) and necrotic area (curved arrow) lack enhancement. (c) Short tau inversion recovery (STIR) coronal sequence shows the tumor to be very heterogenous. Surrounding soft tissue edema appears as bright signal
Bone scintigraphy: Triple-phase, whole-body 99mTc bone scintigraphy can help determine sites of bony metastases, polyostotic involvement, and intraosseous tumor extent. Lesions must be at least 1 cm in diameter to be detected by bone scan. Blood flow, blood pool, and delayed images reveal intense radiotracer uptake. Bone scans may show ossified lymph nodes or calcified lung metastases.
PET-CT: On 18 F-FDG PET-CT, conventional osteosarcoma appears as a highly 18 F-FDG–avid lesion. Tumor aggressiveness correlates with 18 F-FDG uptake, measured by tumor-to-background (T/BG) ratio, SUV, or kinetic modeling techniques [16].
Radiologic differential diagnosis: Ewing sarcoma arises more often in the diaphysis than osteosarcoma. When malignant fibrous histiocytoma arises from bone, it forms a permeative, aggressive metaphyseal lesion that may be difficult to differentiate from lytic osteosarcoma on imaging [17].
Pathologic Features
As current treatment regimens utilize presurgical chemotherapy, it is unusual to encounter a resection specimen that has not been altered by treatment. Grossly, conventional osteosarcomas typically form large destructive tumors that transect the cortex, extend into soft tissue, and contain variable degrees of necrosis, calcification and hemorrhage (Fig. 4.3). Even without presurgical treatment, necrosis may be extensive.
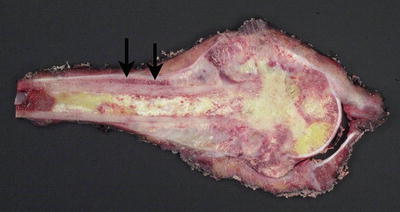
Fig. 4.3
Cut section of osteosarcoma of the femur. The destructive tumor arises from the metaphysis, with extensive involvement of the marrow cavity and extension into soft tissue. Note the elevation of the periosteum (arrows)
Histologically, diagnosis of conventional osteosarcoma requires two fundamental elements: atypical mesenchymal cells and tumoral (as opposed to reactive) osteoid or bone. Osteoid may be quite focal. The prominence of these features varies by patient and by microscopic field, a fact important to consider with small biopsies. Usually, conventional osteosarcoma is a straightforward diagnosis. Most typically, malignant mesenchymal cells are dense and mitotically active, with hyperchromatism, cytologic atypia, and pleomorphism (Fig. 4.4). Tumor cells may be spindled, round, or polymorphic; giant cells, markedly pleomorphic cells (monstrocellular cells), and atypical mitotic figures are common. Osteoid comprises a pink, dense, amorphous material abutting tumor cells, and fibrosis and atypical chondroid matrix are also common. Distinction of osteoid from other types of collagenous matrix can be difficult and subjective. In general, a fibrillar matrix is not osteoid but rather non-osteoidal collagen. Occasionally cartilage may dominate, and demonstration of osteoid production may require careful searching [18]. Tumoral osteoid is often wispy and lace-like but may appear as larger deposits with bone production (Fig. 4.5).
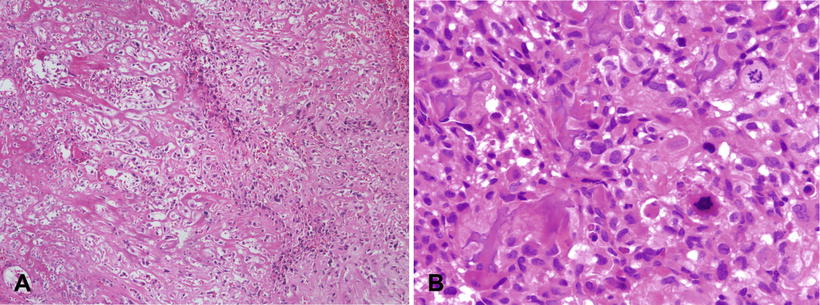
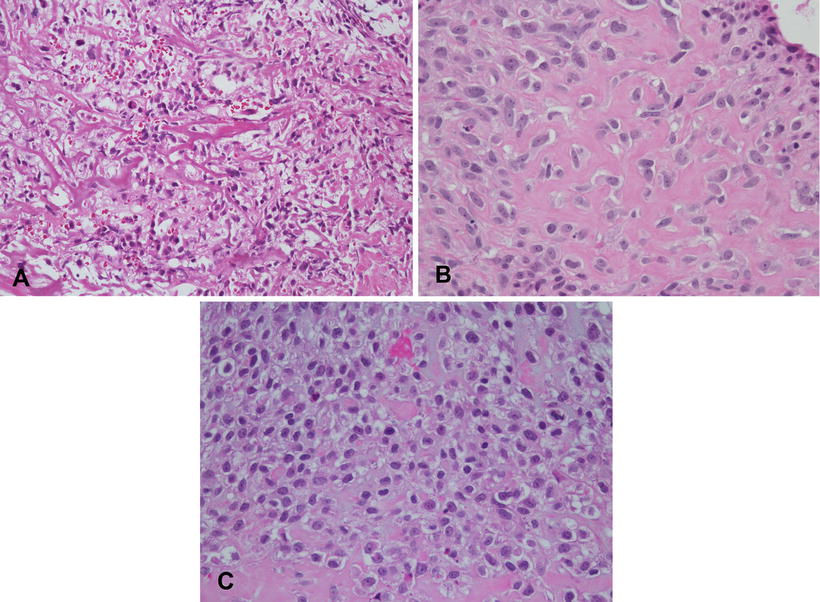
Fig. 4.4
Osteosarcoma, conventional type. (a) Atypical spindle cells enmeshed in lace-like osteoid. (b) High grade features of conventional osteosarcoma; Pleomorphic hyperchromatic spindle cells with atypical mitotic figures. Note the tumoral osteoid in lower left of the figure
Fig. 4.5
Variable appearances of osteoid matrix in osteosarcoma. (a) Lace-like osteoid. (b) Coarser deposits. (c) Non-uniform deposition with subtle lace-like deposits at top of field and coarser deposits at the bottom
In the sclerosing osteosarcoma variant, abundant osteoid matrix totally envelopes tumor cells, giving them the appearance of benign intralacunar osteocytes. The decreased cellularity obscures the malignant nature of the lesion, especially in limited biopsy material [18]. Close attention to the tumor interface with surrounding bone can be helpful in difficult cases, as the leading edges of the tumor often show increased cellularity with atypical cells [18], but such areas may not be present in limited biopsy material. The background reveals variable degrees of hemorrhage, tumoral necrosis and calcification.
Depending upon the predominant matrix component, conventional osteosarcomas have historically been placed into categories of osteoblastic (predominant osteoid and bone production), chondroblastic (predominant chondroid production) or fibroblastic (spindle cells with only focal matrix production) (Fig. 4.6). However, the value of these descriptive categories is debatable, as there can be significant variation in different areas of a given tumor, and moreover, these histologic subtypes appear to have no prognostic significance [9].
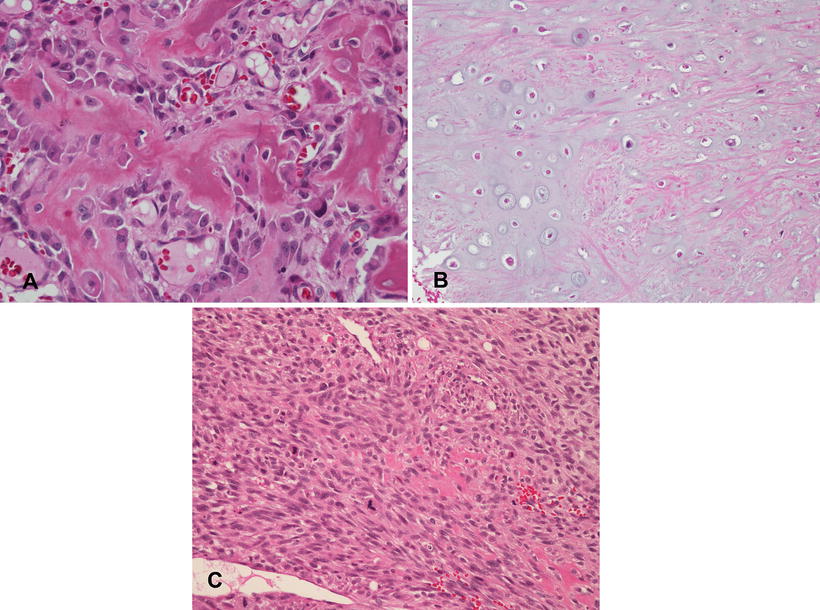
Fig. 4.6
Subtypes of conventional osteosarcoma. (a) Osteoblastic osteosarcoma with “normalization” characterized by solid ribbons of osteoid and minimal nuclear atypia, mimicking an osteoblastoma. Other areas of this tumor showed high grade features (not shown). (b) Chondroblastic osteosarcoma with atypical cartilage. Foci of malignant osteoid production were identified in other areas (not shown). (c) Fibroblastic osteosarcoma. Note the single focus of osteoid production in the center of the field
Rare types of osteosarcoma that present difficulties in diagnosis include epithelioid and osteoblastoma-like tumors. Epithelioid osteosarcoma contains large round to polyhedral undifferentiated cells that resemble epithelial cells and produce little osteoid (Fig. 4.7). They may have prominent nucleoli and on occasion can be cohesive, mimicking an epithelial malignancy or angiosarcoma, so that careful search for osteoid becomes necessary to establish the diagnosis [18]. Cytokeratin positivity makes this distinction even more problematic (see below). Osteoblastoma-like osteosarcoma contains broad bony trabeculae and osteoblastic rimming that mimics osteoblastoma; atypical osteoblasts may also be present. However, it shows infiltration of surrounding bone and atypical mitoses [18].
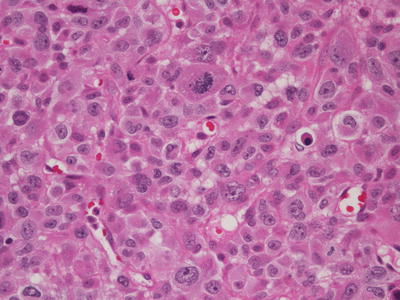
Fig. 4.7
Epithelioid osteosarcoma. Atypical plump cells contain abundant eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and an atypical mitosis. Osteoid production in this tumor (not shown) only became evident following chemotherapy
Immunohistochemical stains: In most cases a firm diagnosis of conventional osteosarcoma only requires routine H&E staining. Immunohistochemistry has limited utility and is mainly used to exclude tumors with better defined and more specific immunohistochemical profiles. The only consistently positive stain in general clinical use is vimentin, and smooth muscle actin may be positive as well [11]. Immunohistochemistry is most helpful in biopsies of high grade primary bone tumors with cellular pleomorphism, necrosis, and hemorrhage, but lacking tumoral osteoid or bone. Such tumors usually represent osteosarcoma, as few other primary bone tumors of childhood have these features [19] CD99 is helpful in ruling out Ewing sarcoma, as the staining pattern in osteosarcoma tends to be cytoplasmic and granular, as opposed to membranous. Rarely osteosarcomas stain with cytokeratin, which can be a pitfall in distinguishing them from synovial sarcoma, angiosarcoma, and metastatic carcinoma [20].
Diagnostic molecular genetics: No specific translocations or mutations define osteosarcoma, so that molecular cytogenetic studies mainly exclude other entities. Osteosarcomas usually exhibit very complex numerical and structural chromosomal aberrations. Some are recurring but none are consistent enough to be helpful in diagnosis [21]. Common abnormalities include loss of chromosomes 9, 13, and 17, gain of chromosome 1, partial loss of 6q, and chromosomal rearrangements of 11, 19, and 20 [22]. Li-Fraumeni syndrome and hereditary retinoblastoma syndrome markedly increase risk for developing osteosarcoma and, respectively, result in loss of RB1 on chromosome 13q and TP53 on chromosome 17p. A significant percentage of sporadic osteosarcomas also show alterations of these tumor suppressor genes [23].
Prognostic and post-treatment issues: According to SEER, the overall 1994–2003 5-year survival rate in children and adolescents was 61.6 %, with distant hematogenous metastases (most commonly lung) or local disease recurrence associated with much poorer survival [9, 24]. Pelvis or vertebral tumors also have adverse prognostic significance [9, 25].
Pathologic examination of the definitive (post-chemotherapy) resection specimen for margin status and degree of necrosis plays an important role in clinical care. As these specimens are often large and require extensive decalcification and fixation, proper handling to ensure good histologic detail and preservation of important margins is not trivial. In our institution, such specimens are handled in the following manner: Upon receipt in the pathology laboratory, the fresh specimen is photographed and X-rayed, and when necessary, orientation is confirmed with surgical personnel. The entire specimen is inked. The specimen is then frozen at −70 °C. Depending on tumor location, the frozen specimen is sequentially sectioned along the longitudinal axis most likely to demonstrate the greatest tumor area. We obtain band saw sections at approximately 5 mm intervals, usually yielding up to 25–30 slabs. The slabs are then fixed and decalcified. Afterwards one or more slabs having the greatest tumor area is mapped, photocopied, and completely submitted along with bone and soft tissue margins (Fig. 4.8). Using the grid map, the pathologist examines each block and evaluates necrosis, expressed as a percentage of the total tumor surface area. Positive margins at time of definitive surgery are associated with a poor prognosis [24]. With effective chemotherapy, extirpation of malignant cells in osteoblastic and chondroblastic areas leaves a background of acellular matrix, often containing empty lacunae or pyknotic “ghost” cells (Fig. 4.9). Fibroblastic areas tend to be replaced by bland granulation and myxoid tissue (Fig. 4.10) [26]. Using current COG protocols, patients are stratified to either “favorable” or “standard” responses, the forming having necrosis >90 %. Unfortunately, such assessments involve a high level of subjectivity. Large areas of the tumor bed composed of either sheets of tumor cells or expanses of acellular matrix are usually straightforward. However, post-chemotherapy specimens that contain foci of chemoresistant cells are typically less cellular than before therapy. Scattered pleomorphic cells with smudgy nuclei (considered to be viable for purposes of grading) [27] often persist in microscopic fields that are otherwise necrotic. In these situations, assignment of a particular percentage necrosis becomes inherently imprecise, and the literature gives little practical guidance. For additional information, the reader is advised to consult the excellent reference by Raymond et al. [28].
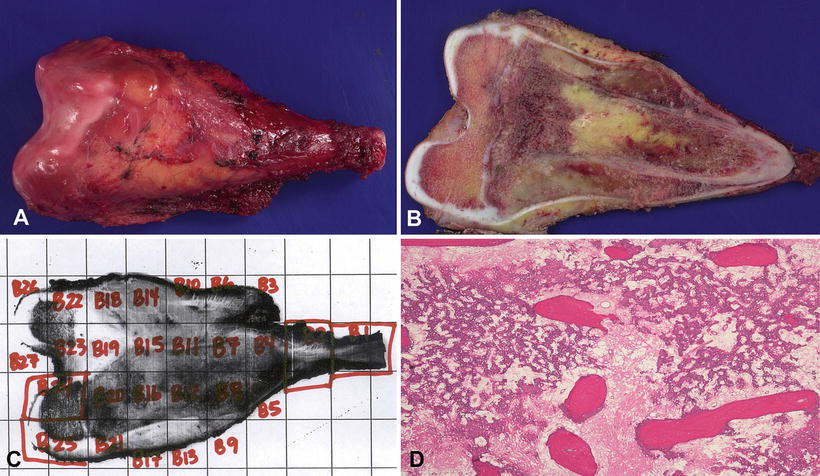
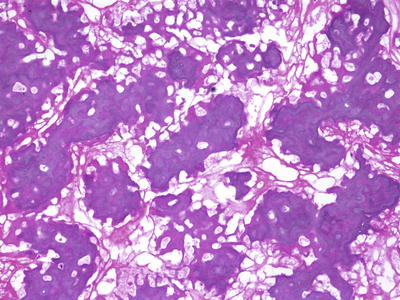
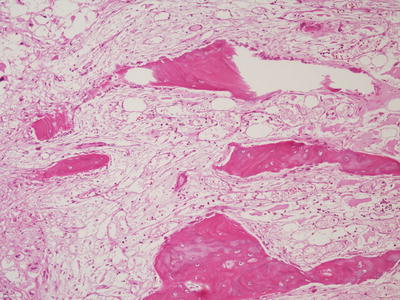
Fig. 4.8
Osteosarcoma, management of post-chemotherapy specimen. (a) Limb-salvage resection of distal femur tumor. (b) Appearance of central tumor slab following freezing of the specimen; note soft tissue and medullary components of the treated tumor. (c) Grid map of the tumor slab, with each labeled rectangle associated with a unique block taken for histologic examination. (d) A microscopic section taken from one of the labeled blocks, demonstrating complete tumor necrosis
Fig. 4.9
Osteosarcoma, post treatment. There is a favorable tumor response with a residual meshwork of acellular osteoid
Fig. 4.10
Osteosarcoma, post treatment: Favorable tumor response with loose myxoid tissue occupying the site of the previous tumor bed. Residual atypical tumor cells are not present
Small Cell Osteosarcoma
Small cell osteosarcoma is rare, accounting for less than 2 % of osteosarcomas. These lesions are small blue cell tumors that demonstrate osteoid production (sometimes focal) but otherwise have features suggestive of Ewing sarcoma. Like conventional pediatric osteosarcoma, it usually arises in long bones and most commonly occurs in the second decade. It may have a slightly worse prognosis than conventional osteosarcoma, although its relative infrequency and controversial diagnostic criteria have raised questions about its behavior.
Imaging Features
Small cell osteosarcoma is an intramedullary, permeative lytic lesion with ill-defined margins that is associated with cortical destruction, aggressive periosteal reaction, and a soft tissue mass [29].
Pathologic Features
Small cell osteosarcoma is composed of sheets of undifferentiated small round cells with hyperchromatic nuclei and minimal pleomorphism, similar to Ewing sarcoma (Fig. 4.11). At least focal osteoid production must be identified to establish the diagnosis [30]. Differential diagnosis can be problematic, as tumoral osteoid may be difficult to separate from the non-osteoidal matrix that may be seen in Ewing sarcoma. Tumor cell spindling favors a diagnosis of osteosarcoma, as does lack of strong, perimembranous CD99 staining. However, Ewing sarcomas may occasionally demonstrate areas of mild spindling, and CD99 positivity has also been described in small cell osteosarcoma, further complicating distinction between the two entities [31]. Most, if not all small cell osteosarcomas lack fusion transcripts for EWSR1-FLI1 or EWSR1-ERG, and although the matter is not entirely settled, some investigators have argued that small cell tumors which harbor these translocations should be considered Ewing sarcomas, regardless of whether or not they produce osteoid [18].
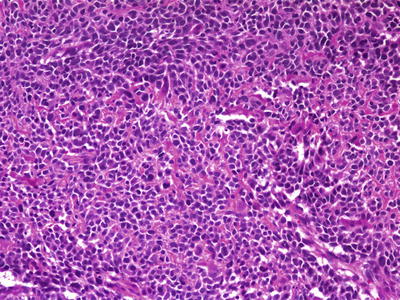
Fig. 4.11
Small cell osteosarcoma. Undifferentiated tumor cells with hyperchromatism and minimal cytoplasm, mimicking Ewing sarcoma. Subtle tumoral osteoid matrix is present in the background
Telangiectatic Osteosarcoma
Telangiectatic osteosarcoma is a rare (4 % of osteosarcomas) high grade subtype of osteosarcoma, with similar demographics regarding age and tumor location [32]. Approximately a quarter of patients present with pathologic fracture. Originally thought to have a worse prognosis than conventional osteosarcoma, survival figures with modern chemotherapeutic regimens are now comparable [33].
Imaging Features
Plain-film radiography: Classic telangiectatic osteosarcoma appears as a geographic area of bone destruction with endosteal scalloping, a wide zone of transition, and marked aneurysmal bony expansion. Its radiographic appearance mimics that of an aneurysmal bone cyst (Fig. 4.12a, b). Osteoid matrix may be subtly visible as small foci in the periphery of the lesion, but most of the lesion is hemorrhagic or necrotic, with multiple hemorrhagic fluid levels [33–36]. Identification of matrix precludes other differential diagnoses such as Ewing sarcoma, aneurysmal bone cyst, and lymphoma [36]. In the 40 patients with telangiectatic osteosarcoma analyzed by Murphey et al. [34], subtle radiographic findings included cortical interruption (78 %), soft tissue mass (19 %), and aggressive periosteal reaction (72 %).
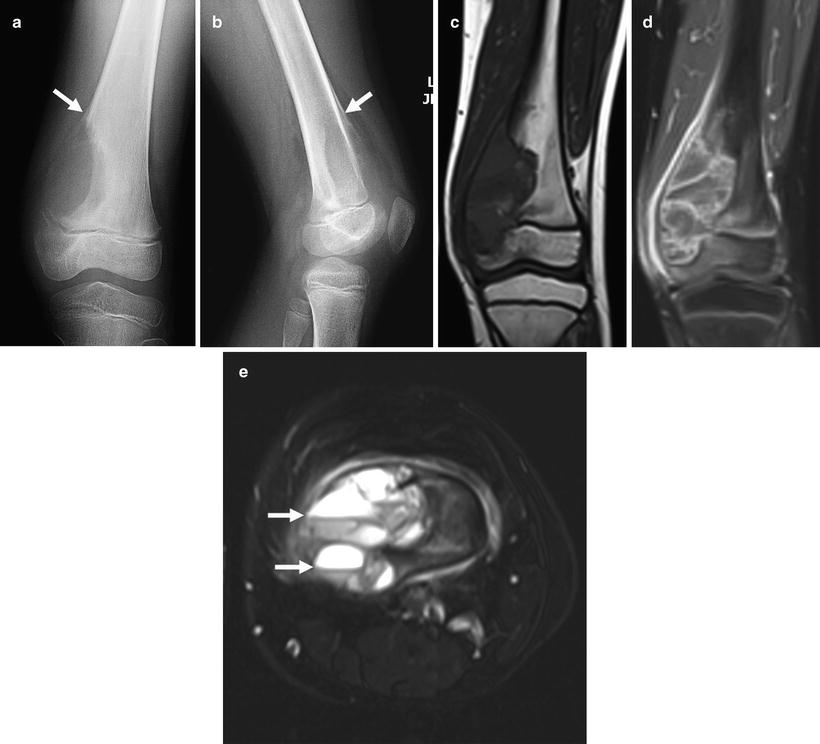
Fig. 4.12
Eight year old girl with telangiectatic osteosarcoma of the distal femur. (a) AP and (b) lateral X-rays show an expansile lucent lesion and Codman triangle (arrows) indicating aggressiveness. Note cortical destruction, also a sign of aggressiveness. (c) T1W coronal MRI shows the expansile lesion invading the distal epiphysis. The bright foci within the tumor are likely due to blood products. (d) Coronal contrast-enhanced T1W MR image showing numerous rim enhancing cystic spaces typical of telangiectatic osteosarcoma. (e) T2W axial MR image shows numerous fluid-fluid levels (arrows) within cystic spaces
Computed tomography: CT typically shows heterogeneous areas of low attenuation in the center of a soft tissue mass that replaces the normal medullary cavity. Cortical interruption and extension of the mass into the adjacent soft tissues is better visualized on CT than on plain films. In a retrospective CT study of 40 patients with telangiectatic osteosarcoma, fluid levels occurred in 48 % and osteoid matrix in 85 % of cases [34]. Post-contrast tumoral enhancement may be seen in the septa surrounding blood cavities. On CT, an intraosseous or soft-tissue osteoid matrix within nodular or septal regions helps distinguish telangiectatic osteosarcoma from aneurysmal bone cyst. Telangiectatic osteosarcoma is associated with aggressive growth features, i.e., cortical destruction and soft tissue extension. In contrast, aneurysmal bone cysts cause marked expansile remodeling of bone and cortical thinning but lack true soft tissue involvement [34].
Magnetic resonance imaging: MRI is the best modality to evaluate the soft-tissue component. Generally, telangiectatic osteosarcoma has heterogeneous signal intensity on both T1W and T2W images (Fig. 4.12c–e). T1W images show isointense or hyperintense signal to skeletal muscle [33, 36]. Low signal on T1W may represent calcifications. T2W images are ideal to demonstrate associated fluid levels. In a study by Murphey et al. fluid levels were more often detected by MRI (89 %) than CT and represented hemorrhage [34]. Postgadolinium-enhanced images show peripheral nodular enhancement [36], which helps to distinguish this neoplasm from aneurysmal bone cyst, which has thin peripheral septa (usually 2–3 mm thickness) often seen as enhancing structures that lack nodularity.
Bone scintigraphy: The typical bone scintigraphic finding of telangiectatic osteosarcoma is the “doughnut sign,” which is due to peripheral uptake in the solid portions of the lesion and central photopenic areas in the fluid components [34].
Pathologic features
The gross appearance of telangiectatic osteosarcoma is similar to aneurysmal bone cyst, with medullary based grossly apparent cysts containing blood and blood clots and lacking a fleshy or fibrotic component (Fig. 4.13). Histologically, the architectural resemblance to aneurysmal bone cyst is maintained, with fibrous septa of variable sizes surrounding zones of hemorrhage and blood clot. Septa are often lined by bland spindle and giant cells, but unlike aneurysmal bone cysts they usually contain conspicuous anaplastic, hyperchromatic, mitotically active, and markedly pleomorphic cells. These cells produce variable amounts of osteoid, often lace-like and limited in amount (Fig. 4.14a, b). Osteoclast-like giant cells may be numerous [18].
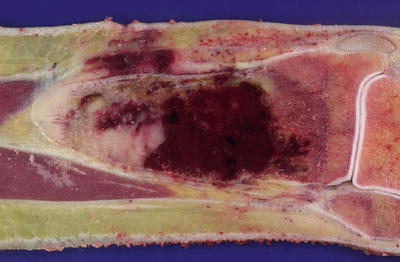
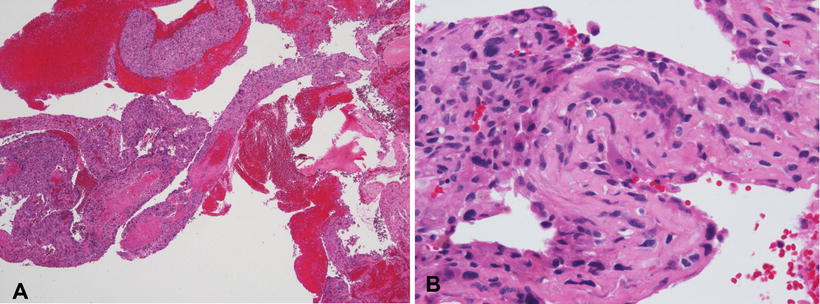
Fig. 4.13
Telangiectatic osteosarcoma arising in distal tibia. Tumor is extensively hemorrhagic and extends into soft tissue
Fig. 4.14
Telangiectatic osteosarcoma. (a) Membranous septa admixed with blood. (b) Membranous septum with atypical tumor cells (left) and osteoclast-like giant cells (right)
Low-grade Central Osteosarcoma
Low-grade central osteosarcoma accounts for less than 2 % of osteosarcomas and has a favorable prognosis with complete surgical resection [37]. It only occurs in long bones. Histologically, it resembles parosteal osteosarcoma, with bony trabeculae separated by a hypocellular fibrous stroma containing minimally to moderately atypical spindle cells. Mitotic activity is always present but may be low. Cytologic atypia, infiltration, and permeation distinguish low-grade central osteosarcoma from benign processes such as fibrous dysplasia and chondromyxoid fibroma [37]. Few studies have investigated the molecular genetics of this rare neoplasm. In one study of six patients, comparative genomic hybridization showed recurrent gains at 12q13-14, showing genetic as well as histologic relatedness to parosteal osteosarcoma [38].
Parosteal Osteosarcoma
Surface osteosarcomas arise from the periosteum or cortex, with minimal or no involvement of the underlying medullary cavity. The three major types are parosteal osteosarcoma, periosteal osteosarcoma, and high grade surface osteosarcoma. Parosteal osteosarcoma is a low grade tumor that accounts for 4 % of osteosarcomas in general but is rare in childhood. Most cases occur in young adults [39]. The most common site is the distal posterior femur, with occasional examples in other long bones. Origin in flat bones is exceptional. With total resection, the prognosis of these slow growing tumors is excellent, although there is a 10 % risk of distant metastatic disease [18]. Unlike conventional osteosarcoma, these tumors tend to be painless and usually only have symptoms of a mass or restriction of movement [18].
Imaging Features
Plain-film radiography: Parosteal osteosarcoma has an osseous fusiform configuration along the length of the bone. Its tendency to wrap around the bony circumference as it enlarges creates a cauliflower-like appearance contiguous with the cortical surface (Fig. 4.15a) [40, 41]. It may be separated from the underlying cortical bone by a radiolucent cleft. Cortical thickening without aggressive periosteal reaction may also be seen. The bone is more mature at the center of the lesion than the periphery.
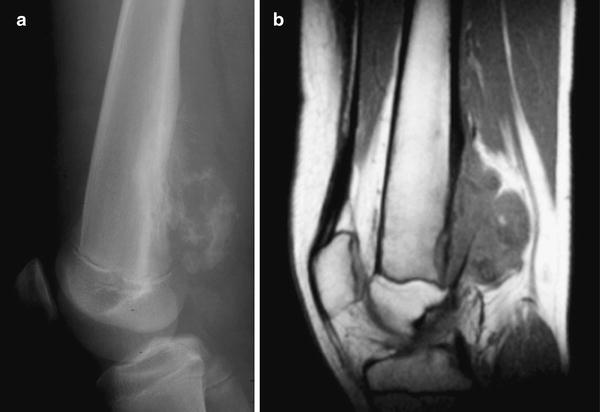
Fig. 4.15
Parosteal osteosarcoma of the distal right femur in a 14 year old boy. (a) Lateral radiograph demonstrates a lobulated, exophytic calcified soft-tissue mass arising from the surface of the distal femur. The radiographic appearance and anatomic location are typical of parosteal osteosarcoma. (b) Sagittal non-contrast-enhanced MR images shows the lobulated soft tissue mass causing cortical erosion and thickening without involvement of the medullary canal
Computed tomography: CT features mimic those of radiography but are more sensitive at identifying occasional soft tissue involvement. CT may also determine bone maturity at the center of lesion versus the periphery. Most parosteal osteosarcomas are low grade, but high grade foci may appear on CT or MRI as non-mineralized soft tissue components with or without fluid levels, measuring larger than 1 cm [40]. Such foci represent dedifferentiation into telangiectatic osteosarcoma, spindle cell sarcoma, or high-grade surface osteosarcoma, which have prognoses similar to that of conventional osteosarcoma.
Magnetic resonance imaging: MRI reveals a heterogeneous appearance with low-signal tumoral bone and high-signal peripheral soft tissue (Fig. 4.15b). Involvement of the cortex and marrow causes a high signal on fluid sensitive sequences and may enhance with contrast [15, 42].
99m Tc MDP scan: Elevated radiotracer activity within the tumor reflects the biological behavior of the tumor and may be valuable in detecting synchronous lesions or metastasis.
Radiographic differential diagnosis: Myositis ossificans has denser peripheral ossification than parosteal osteosarcoma and is not attached to the cortex. In addition, the bone in myositis ossificans is more mature at the periphery than the center. Continuity of cortex and marrow into the stalk of the osteochondroma differentiates it from parosteal osteosarcoma.
Pathologic and Genetic Features
Grossly, parosteal osteosarcomas form lobulated, hard masses fixed to the underlying cortex. A minor degree of marrow invasion may be seen in up to 25 % of cases. Histologically, these tumors contain well formed parallel bony trabeculae immersed in a hypocellular, collagenous stroma (Fig. 4.16). These bony trabeculae mimic normal bone and sometimes have osteoblastic rimming. A cartilage cap may also be present, mimicking a sessile osteochondroma (Fig. 4.17). Unlike conventional osteosarcoma, stromal spindle cells show only minimal to moderate cytologic atypia, often with deceptively bland elongated nuclei and few mitoses. Up to one half of these tumors demonstrate cartilaginous differentiation. High grade spindle cell areas, referred to as dedifferentiation, occur in 15 % of cases. Dedifferentiation more commonly occurs in recurrent lesions and has a prognosis similar to conventional osteosarcoma [43].
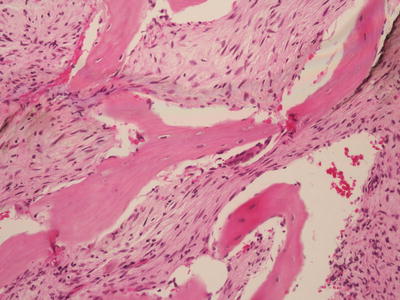
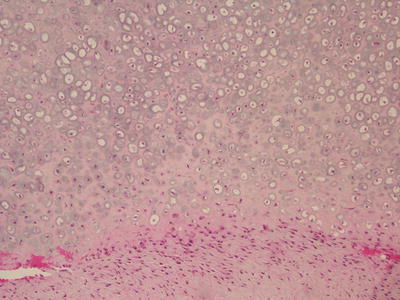
Fig. 4.16
Parosteal osteosarcoma. Parallel oriented trabeculae of woven bone lacking osteoblastic rimming, with a moderately cellular fibrotic background
Fig. 4.17
Parosteal osteosarcoma. This section of peripheral cartilaginous cap consists of thick, low grade atypical cartilage
Differential diagnosis includes low grade central osteosarcoma, osteochondroma, and parosteal osteoma. Central osteosarcomas have much greater medullary involvement. Cartilage caps are common in parosteal osteosarcomas, creating confusion with osteochondromas. However, osteochondromas lack the collagenous spindle cell stroma of parosteal osteosarcomas but instead resemble normal growing bone with fatty or hematopoietic marrow [18]. Correlation with imaging findings is imperative, as evidence of cortical-medullary continuity strongly favors osteochondroma. Parosteal osteomas occasionally involve the long bones, but unlike parosteal osteochondromas they lack spindle cell proliferation [44].
As opposed to conventional osteosarcomas, parosteal osteosarcomas do not display a wide variety of chromosomal gains and losses. Instead, the most common aberration is the presence of one or more supernumerary ring chromosomes, with amplification of 12q13-15 [39].
Periosteal Osteosarcoma
Periosteal osteosarcoma is an intermediate grade surface osteosarcoma characterized by its mainly cartilaginous matrix. Less common than parosteal osteosarcoma, it accounts for fewer than 2 % of all osteosarcomas. Most cases occur in adolescents and young adults. The metaphyseal–diaphyseal regions of long bones such as the tibia and femur are most commonly involved [45], but involvement of the flat bones is rare. Disease-free survival after adequate treatment is >80 %, with most recurrences and deaths occurring within 3 years of diagnosis [46].
Imaging Features
Plain-film radiography: Periosteal osteosarcoma forms a broad-based soft tissue mass causing extrinsic erosion of a thickened underlying diaphyseal cortex. A perpendicular sunburst-type periosteal reaction extends into the soft tissue component, typically without intramedullary involvement (Fig. 4.18a) [47]. The zoning pattern is typical of surface osteosarcoma, with more mature and organized bone at the center than the periphery. The cortex appears variegated and may be thickened, scalloped, or irregular and permeative. There is no true aggressive periosteal reaction, but cortical thickening may resemble the periosteal elevation of Codman’s triangle. Because these lesions are chondroblastic, matrix ossification is not prominent [47].
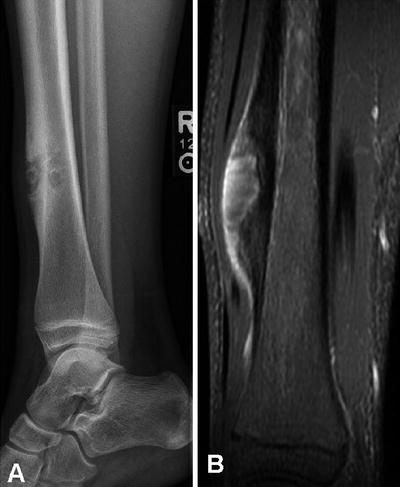
Fig. 4.18
Periosteal osteosarcoma of the distal tibial diaphysis in a 15 year old male. (a) Lateral radiographic view of the distal tibia showing a broad based mass causing erosion of the thickened tibial diaphyseal cortex. (b) T1W fat-saturated post contrast MRI image shows the thickened cortex to be hypointense. There is some enhancement of the soft tissue component and a sunburst type periosteal pattern is present. No intramedullary extension of the tumor is seen
Computed tomography: CT findings mimic radiographic ones, although CT better delineates cartilaginous areas, soft tissue involvement, and zoning of bone maturity. Perpendicular periosteal reaction forms low-attenuation rays.
Magnetic resonance imaging: MRI is useful in evaluating the true extent of the tumor. Cartilage and cortex appear as low signal on T1W and as high signal on T2W sequences (Fig. 4.18b). Low-signal osteoid matrix appears as rays perpendicular to the cortex. High signal contrast enhancement of the cortex or the marrow indicates tumoral involvement of these regions [47].
Pathologic Findings
Grossly, periosteal osteosarcoma arises from the bony surface as a fusiform or rounded mass that may partly or entirely involve the circumference (Fig. 4.19). A sunburst appearance results from calcified spicules that arise from the cortical surface. Glistening white-gray cartilaginous differentiation is usually prominent. Histologically, chondroblastic areas predominate and contain anaplastic spindle cells with focal enchondral ossification (Fig. 4.20) unlike periosteal chondroma. Diagnostic tumoral osteoid must be present but may be relatively inconspicuous, similar to intermediate grade chondroblastic osteosarcoma. The juxtacortical portion of the tumor is usually densely ossified. Whether or not tumor can extend into the underlying medullary cavity is controversial. Some contend it is common [45], while others feel that medullary involvement indicates conventional chondroblastic osteosarcoma with cortical destruction and periosteal spread [46]. The evidence suggests that predominantly surface chondroblastic osteosarcomas with medullary involvement show more aggressive clinical behavior [45].
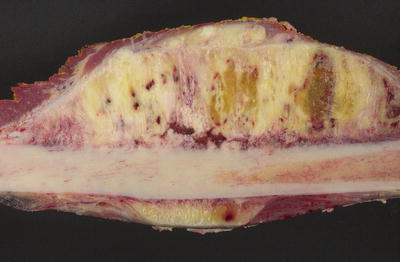
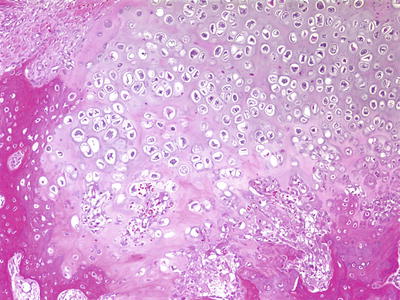
Fig. 4.19
Periosteal osteosarcoma. Bisected femoral shaft demonstrating surface location and sunburst appearance
Fig. 4.20
Periosteal osteosarcoma. Lobule of malignant cartilage with associated bone formation
High-Grade Surface Osteosarcoma
High grade surface osteosarcoma is a high grade osteosarcoma arising from the surface of the bone. Extremely rare (<1 % of all osteosarcomas), it has the clinical presentation, demographic distribution, and prognosis of conventional osteosarcoma. The most common site of involvement is the midshaft of the femur [48].
Imaging Features
Plain-film radiography: High-grade surface osteosarcoma appears as an osteoid matrix present in a soft tissue mass. Distortion of fat planes around the soft tissue mass and scalloping or permeative destruction of the underlying cortex may occur [15, 49]. Periosteal reaction is common and tends to be aggressive. High grade surface osteosarcomas typically involve the entire circumference of the bone and may involve the medullary canal.
Computed tomography: CT findings mimic radiographic ones and improve visualization of the osteoid matrix and soft tissue involvement.
Magnetic resonance imaging: MRI shows a soft tissue mass containing variable amounts of low-signal osteoid matrix, giving an overall inhomogeneous signal on fluid-sensitive sequences. Periosteal reaction and surrounding edema in the adjacent cortex are visible. Soft tissue enhances avidly on contrast administration.
Pathologic findings
Grossly, high grade surface osteosarcomas are attached to the underlying cortex and frequently have a variegated appearance imparted by intermixed zones of hard matrix production and softer hemorrhage and necrosis. The latter soft areas are not typical of lower grade surface tumors [50]. Histologically, these tumors are indistinguishable from conventional osteosarcomas and have chondroblastic, fibroblastic, and osteoblastic variants [48, 51]. If significant medullary involvement is present, there is a possibility that the lesion originated from a conventional osteosarcoma that broke through the cortex and developed a dominant surface mass. However, both are high grade lesions treated in a similar fashion.
Ewing Sarcoma
Ewing sarcoma (ES) is the second most common primary malignancy of bone in childhood. Although first described as a primary undifferentiated bone tumor, it also occurs in soft tissue and may exhibit variable degrees of neuroectodermal differentiation, designated “primitive neuroectodermal tumor” (PNET). Ewing sarcomas and PNETs involving either bone or soft tissue share identical recurrent translocations. No longer biologically distinct, these entities have now been subsumed into the category of “Ewing’s sarcoma family of tumors” (ESFT’s) [52]. ESFTs are most common in whites, rare in African and African-American children, and slightly more common in males than females [53]. They most commonly affect adolescents but occur at any age, even infancy [54]. Usually they arise from the diaphysis of long bones or the pelvis, but any bone can be involved. [55]. ESFTs are clinically aggressive, and prior to the advent of chemotherapy nearly all patients died of their disease. With current treatment regimens, the overall survival of patients with localized disease is 75 %. However, one quarter of patients have metastatic disease at diagnosis and a poor prognosis [56]. Tumors commonly metastasize to lung; other sites include bone marrow and other bones. Although pain with or without pathologic fracture is the most common presenting symptom, patients may show signs and symptoms mimicking infection [55].
Imaging Features
Plain-film radiography: The tumor originates in the medullary cavity, invades the Haversian system, and extends into the subperiosteal areas (Fig. 4.21a). In a study of 64 patients with Ewing sarcoma (ES), Peersman et al. found that a mixed sclerotic-lytic pattern of permeative lytic cortical destruction and a wide zone of transition were the most common patterns (40 %) [57]. Reinus et al. found cortical thickening in 58 of 274 (21 %) patients with ES [58]. Cortical violation and a soft tissue mass are frequently seen. Extension into the subperiosteal region can cause an aggressive lamellated or sunburst periosteal reaction and a Codman’s triangle. The tumor cannot produce new bone or cartilaginous matrix, but reactive bone is formed. Soft tissue extension of the tumor will not contain bone [59, 60].
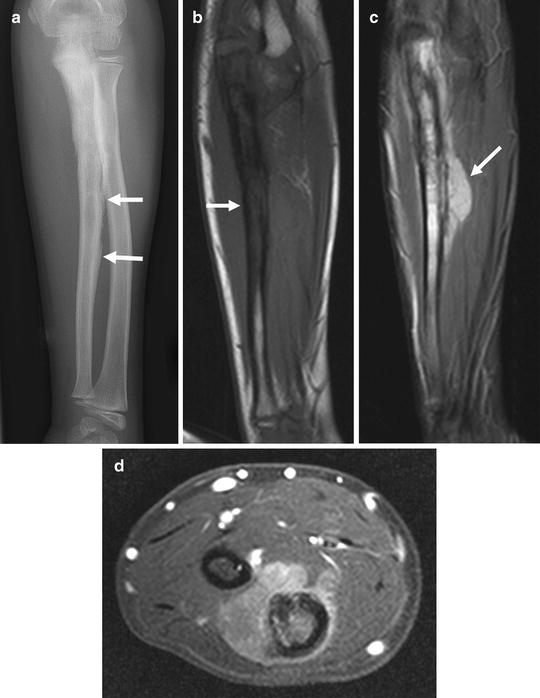
Fig. 4.21
Ten year old boy with Ewing sarcoma of the ulna. (a) AP radiograph shows a permeative, medullary process with wide zone of transition in the proximal and mid ulna. Note the lamellar, irregular, periosteal reaction (arrows) typical of Ewing sarcoma. (b) Coronal TIW MR image demonstrates the medullary tumor as hypointense to muscle (arrow). (c) On STIR MR imaging the tumor is hyperintense and the soft tissue component (a typical feature of Ewing sarcoma) is evident (arrow). (d) Post-contrast axial MR image shows moderate enhancement of the medullary and soft-tissue components of the tumor
Computed Tomography: CT shows cortical disruption and soft tissue extension more clearly than plain films. Chest CT is important to detect lung metastases [61].
Magnetic resonance imaging: MRI is more sensitive than CT in detecting bone marrow and soft tissue infiltration, and aids in pinpointing the site for bone biopsy [57, 62]. MRI can show cortical tumoral involvement because of its prolonged T1 and T2 relaxation times [57]. MRI also shows skip lesions. The tumor appears isointense or hypointense to skeletal muscle on T1W images and homogenously hyperintense on T2W images (Fig. 4.21b). Reactive bone may form areas of low signal within the marrow. Any associated marrow or soft tissue edema or periosteal reaction appears as a high signal on STIR sequences (Fig. 4.21c). Post-contrast images show avid but inhomogeneous enhancement (Fig. 4.21d). Low signal central areas within the tumor may represent necrosis. Dynamic contrast enhanced MRI or diffusion-weighted MRI may indicate the extent of tumor necrosis [63]. MRI is also used for restaging and evaluating response to treatment. Signs of response to therapy include a decrease in tumor size, maturation of the periosteal reaction, and bony sclerosis [63].
Positron emission tomography: An increased rate of glycolysis in ES causes increased FDG avidity, thereby facilitating tumor detection and making the biologically active tumor more conspicuous than the surrounding tissue. Hawkins et al. [64] found that the mean standardized uptake value (SUV) for ES was approximately 5.3 (n = 14). The SUV of ES, both before and after neoadjuvant therapy, predicts patient outcome [64–67]. In addition, qualitative evaluation of FDG accumulation within a large heterogeneous tumor helps identify active areas most suitable for targeted biopsies.
FDG PET is used in staging ES, as it is more sensitive than a bone scan (88 % vs. 37 %) for detecting bone marrow disease and the osteoclast-mediated response to osseous metastasis [65]. As biochemical changes in the tumor precede morphological changes [57, 68], PET-CT can often indicate tumoral regression or progression earlier than CT and MRI. Also, FDG PET is superior to bone scintigraphy in assessing the chemotherapy response of bone metastases [66].
Bone scintigraphy: Scintigraphy with 99mTc-MDP shows increased uptake on a three-phase bone scan (angiographic, blood pool, and delayed). However, bone scan can miss very small degrees of metastasis or bone marrow disease and is not sensitive enough to detect the local extent of disease or skip metastasis. Metastases appear as scattered foci of increased uptake in axial skeleton with red marrow. Photopenic foci due to necrosis often occur with aggressive lesions.
Pathologic Features and Differential Diagnosis
As with osteosarcoma, gross specimens of Ewing sarcoma are generally altered by presurgical treatment. Although the tumor may be localized to bone, invasion into surrounding soft tissues is frequent and often massive, masking exact origin of tumor from soft tissue or bone (particularly those in chest wall) (Fig. 4.22). By mapping slab grids of decalcified tumor similar to osteosarcoma, chemosensitivity can be evaluated and correlates with patient outcome [69, 70]. However, the inherent subjectivity in such assessments is magnified by the tendency of Ewing tumor beds to shrink as a response to treatment. Unlike osteosarcomas, Ewing sarcomas do not leave behind devitalized matrix, so that determining tumor boundaries for evaluating tumor necrosis can be quite difficult, raising questions as to the reproducibility of such efforts.
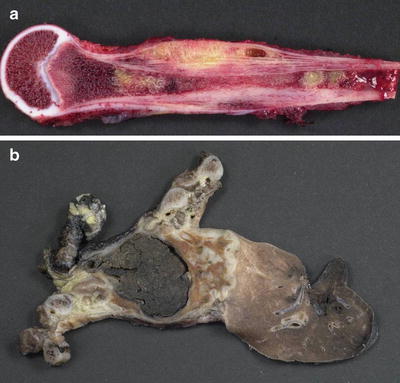
Fig. 4.22
Ewing sarcoma. (a) Bisected humerus with diaphyseal tumor and soft tissue extension. (b) Chest wall resection and right lung lobectomy (fixed and post-treatment) with extensively hemorrhagic and necrotic tumor. This case demonstrates the inherent difficulty in determining origin from a rib or from the adjacent soft tissues of the chest wall
Histologically, Ewing sarcoma can be considered the prototype of the “small round blue cell tumor” of childhood. The classic microscopic appearance is of non-matrix producing sheets of uniform undifferentiated small cells with no particular architectural pattern except for occasional rosettes (Fig. 4.23a–d). The typical Ewing sarcoma cell is small (diameter less than twice that of an endothelial cell) and round to oval with minimal to no spindling. It has a high nuclear to cytoplasmic ratio with relatively little cytoplasm and a nucleus with powdery to finely dispersed chromatin. Nucleoli are usually not prominent. Cytoplasm may be cleared or eosinophilic. Most tumors contain cytoplasmic glycogen demonstrable with periodic acid-Schiff (PAS) stains, historically part of the routine diagnostic workup. However, a few lack stainable cytoplasmic glycogen, and many non-Ewing sarcomas contain it. Cytologic pleomorphism is usually minimal, and mitotic activity can be variable. Indeed, many of these tumors display surprisingly low mitotic rates, considering their aggressiveness. Commonly, scanning discloses a mixture of so-called “light” and “dark” cells. The dark cells possibly represent effete cells undergoing degenerative changes and having more hyperchromatic and dense nuclei (Fig. 4.24). Extensive necrosis is not unusual in pre-treatment biopsies.
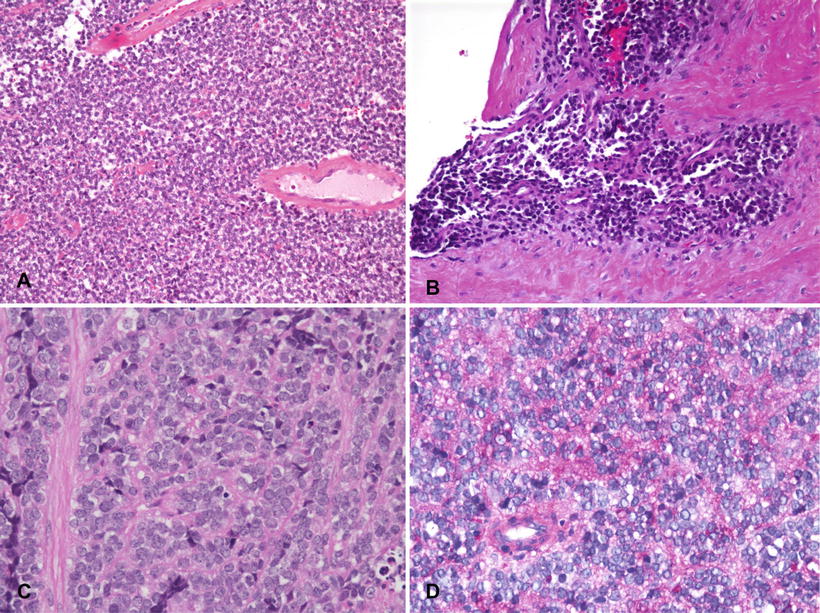
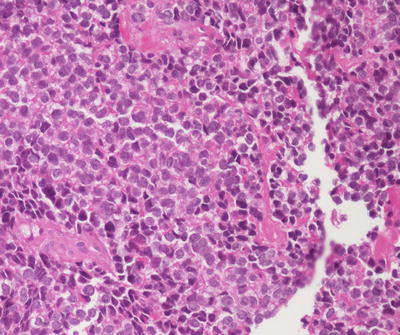
Fig. 4.23
Ewing sarcoma. (a) Typical appearance of sheets of undifferentiated “small round blue cells” with no particular architectural pattern. (b) Invasion into surrounding soft tissue. (c) Typical cytologic features of monotonous round cells with fine chromatin, inconspicuous nucleoli and sparse cytoplasm. (d) PAS stain demonstrating abundant cytoplasmic glycogen
Fig. 4.24
Ewing sarcoma. The lesion contains alternating “light” and “dark” cells
Although the great majority of Ewing sarcomas have “classic” features, variations may cause diagnostic uncertainty (Fig. 4.25a–c). “Adamantinoma-like” Ewing sarcomas display a nested growth pattern with desmoplasia and peripheral palisading akin to true adamantinomas and accounted for 5 % of ESFT in one large series. Rare cases with abundant matrix (sclerosing pattern), extensive spindling reminiscent of synovial sarcoma (spindle cell sarcoma-like pattern), and high mitotic activity, more pleomorphism, nuclear enlargement, and prominent nucleoli (atypical Ewing sarcoma) were also encountered [71]. CD99- and translocation-positive cases with rhabdoid-like, epithelioid, or hemangioendotheliomatous features also occur [72, 73]. These rare cases broaden the morphologic spectrum of Ewing sarcomas and underscore the importance of molecular testing for Ewing associated fusions, but to date have bestowed no prognostic significance [52].
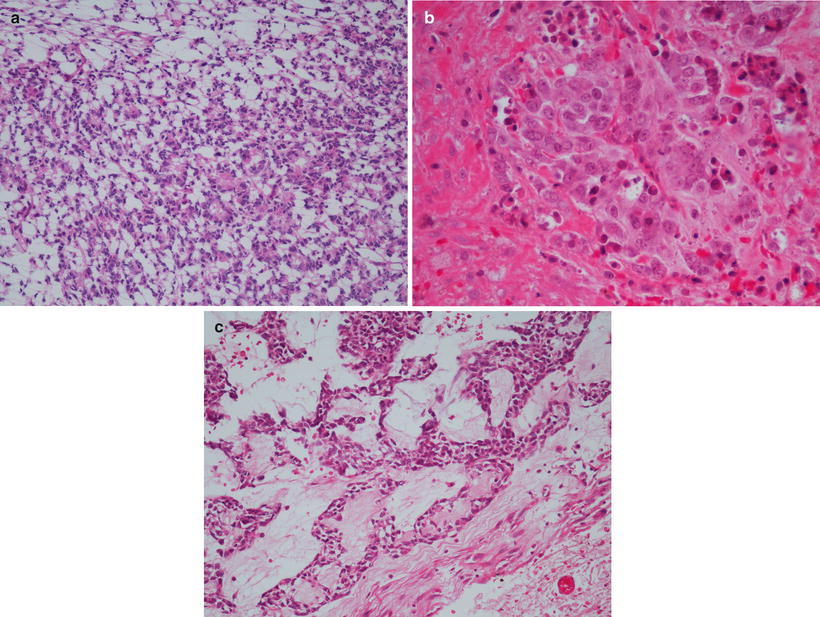
Fig. 4.25
Ewing sarcoma histologic variants. (a) Primitive neuroectodermal tumor pattern with neuroepithelial rosettes. (b) Large cell variant. (c) Adamantinoma-like variant
Immunohistochemically, the vast majority of Ewing sarcomas express cytoplasmic vimentin and caveolin-1 and membranous CD99 (Fig. 4.26) [74]. CD99 is highly sensitive but lacks specificity, as it is also expressed in lymphomas and some rhabdomyosarcomas, mesenchymal chondrosarcomas, rhabdoid tumors, and synovial sarcomas. Nuclear or granular cytoplasmic CD99 expression does not support a diagnosis of Ewing sarcoma. Most (75–91 %) Ewing sarcomas express nuclear FLI-1 [71, 75], a finding that is also not entirely specific and seen in some lymphomas. Up to 20 % of Ewing sarcomas display focal cytokeratin positivity [76], and rare cases with epithelial membrane antigen and desmin positivity have been described [77]. Neuronal markers such as neuron-specific enolase (NSE), synaptophysin, CD56, S-100, and neurofilament protein are variably positive, especially in PNET.
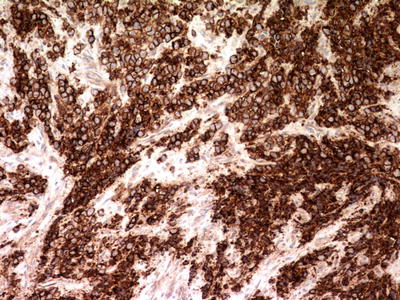
Fig. 4.26
Ewing sarcoma. Immunohistochemistry for CD99 displays strong and uniform perimembranous staining
Ultrastructurally, Ewing sarcoma cells appear undifferentiated, contain few organelles, and show abundant cytoplasmic glycogen. Dense core neurosecretory granules and dendritic processes are occasional, especially in PNET. Unlike lymphomas, Ewing sarcoma cells contain primitive intercellular junctions [55]. This can be a helpful cytological feature, as intraoperative touch imprints typically contain cohesive cells in a clean background and differ from the singly dispersed cells and lymphoglandular bodies of lymphomas.
Given the undifferentiated nature of Ewing sarcoma, differential diagnosis with other undifferentiated malignancies can be challenging and requires judicious application of ancillary techniques. Particularly challenging is its distinction from lymphoblastic lymphoma of bone, metastatic undifferentiated neuroblastoma, metastatic rhabdomyosarcoma, small cell osteosarcoma, and mesenchymal chondrosarcoma. Both Ewing sarcoma and lymphoblastic lymphoma may display perimembranous CD99 positivity and lack CD45 staining, but lymphoblastic lymphoma can be diagnosed by its expression of TdT and more general markers of B- or T-cell lineage. When enough fresh tissue is available, flow cytometry can be very helpful. Small cell osteosarcoma must be diagnosed if tumoral osteoid is present. A recent report suggests that small cell osteosarcomas and mesenchymal chondrosarcomas are negative for FLI-1 [75]. In general, tumors that lack strong perimembranous staining for CD99 should not be considered Ewing sarcoma in the absence of confirmatory molecular data, as only 1 % of proven Ewing sarcomas have been found to lack it [72]. CD99 staining is particularly helpful in ruling out metastatic neuroblastoma, as neuroblastomas should be negative, and likewise myogenin can be used to exclude metastatic rhabdomyosarcoma, as Ewing sarcoma does not express that antigen. Unlike neuroblastoma and rhabdomyosarcoma, Ewing sarcoma is typically (but not always) CD56-negative [78, 79].
Diagnostic molecular genetics: The great majority of Ewing sarcomas harbor EWSR1 translocations, which can be demonstrated with both fluorescence in situ hybridization (FISH) and reverse transcription-polymerase chain reaction (RT-PCR) [80]. FISH has become more commonly used than RT-PCR, as it is widely available, applicable to paraffin embedded formalin fixed tissue, and does not require specific primers for each possible translocation partner. This is of particular importance in Ewing sarcoma, as it has the greatest variety of fusion patterns of any translocation-associated pediatric sarcoma [81].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


