Classification for pediatric renal tumors
Nephroblastic tumors
Nephroblastoma (Wilms’ tumor)
Favorable histology
Anaplasia (diffuse or focal)
Nephrogenic rests and nephroblastomatosis
Cystic nephroma and cystic partially differentiated nephroblastoma
Mesoblastic nephroma
Cellular
Classic
Mixed
Clear cell sarcoma
Rhabdoid tumor
Renal epithelial tumors of childhood
Translocation-associated tumors
Renal cell carcinoma associated with Xp11.2 translocations (TFE3)
Other translocation-associated tumors
Papillary renal cell carcinoma
Renal medullary carcinoma
Oncocytic renal neoplasms after neuroblastoma
Chromophobe renal cell carcinoma
Rare tumors
Primitive neuroectodermal tumors (PNET)
Primary rhabdomyosarcoma of the kidney
Primary synovial sarcoma
Overview of Imaging Features
The imaging evaluation of a suspected pediatric renal mass should always begin with ultrasound. Sonography is readily available, can be performed without sedation and intravenous contrast, and does not expose the child to ionizing radiation. If a renal mass is identified on ultrasound, further evaluation of the renal vessels and inferior vena cava (IVC) with both gray scale and color Doppler images are required to detect vascular extension, present in up to 10 % of cases [4]. Further evaluation with contrast enhanced computed tomography (CT) or magnetic resonance imaging (MRI) is also required for local staging of the mass prior to any intervention, as per the guidelines of the Children’s Oncology Group (COG). Either modality can be used, depending on institutional availability and expertise [5].
In keeping with the As Low As Reasonably Achievable (ALARA) principle regarding ionizing radiation exposure, pre-contrast and multiphase contrast enhanced CT images are not required for diagnosis or staging of pediatric renal tumors [6]. A single-phase study in the portal venous phase (approximately 50 s after contrast injection) sufficiently stages a renal tumor and delineates relevant anatomy. Multiplanar CT reconstructions or multiplanar scanning with MRI can confirm the renal origin of the mass and assess its relationship to vital structures such as the renal vessels. If a partial nephrectomy is being considered, multiphase images are helpful to determine the relationship of the mass to the renal vessels and collecting system. A CT of the chest is required in all malignant renal tumors to evaluate for potential lung metastasis.
Wilm’s Tumor
Definition
Wilms’ tumor (syn: nephroblastoma), a malignant neoplasm originating from nephrogenic blastemal cells, is the second most common malignant, solid extracranial tumor in children [1, 7, 8]. It is a traditional blastematous tumor, exhibiting various stages of embryonic development and multiple lines of differentiation.
Clinical Features and Epidemiology
In children ranging from 0 to 15 years of age, Wilms’ tumor occurs in seven to ten cases per million annually (1 in 10,000 children) and accounts for approximately 95 % of renal tumors, and comprises 6–7 % of all pediatric tumors [7, 9, 10]. It is the fifth most common pediatric tumor and the second most common intraabdominal tumor in children [11].
Wilms’ tumor predominantly arises in young children. Greater than 80 % of Wilms’ tumors are found in children younger than 5 years old, with an average age of 3.5 years [1]. Rarely, 0.16 % of Wilms’ tumors are seen in neonates, and on very few occasions it occurs in adults [7].
Wilms’ tumor statistics vary with respect to ethnicities. In the USA, there is a lower incidence in Hispanic/Latino compared with non-Hispanic children (RR = 0.78, 95 % confidence interval = [0.64, 0.95]) [9]. The incidences in Chinese and American black children are 2.5 per million and 10.9 per million, respectively [7]. In Britain, the incidence is lower among Asian children than Caucasian children (RR = 0.51, p < 0.05) but higher among West Indian children (RR = 2.55, p < 0.05) [9].
Wilms’ tumor has a slightly greater propensity to occur in girls (9.7 per million) than boys (8.4 per million). Data has shown that the age at diagnosis is significantly higher for girls than boys. The Wilms’ tumor population in Europe shows similar trends with a 0.9 ratio of boys to girls (median age for girls 3 years; for boys 2 years). In Asia, a higher reported percentage of boys developed Wilms’ tumor [9].
More than 90 % of Wilms’ tumors present as an asymptomatic abdominal mass. Most are contained within a single lesion, 6 % present as bilateral tumors, and 12 % have multifocal disease within a single kidney. If abdominal pain (20–40 %) exists at diagnosis, there is a risk of rupture and bleeding [1, 12]. Gross hematuria (5–25 %) indicates tumor invasion into the collecting system or ureter [1, 12]. Rare cases have been reported at extrarenal sites, such as in the perirenal, inguinal, or gonadal areas, perhaps as a component of “monodermal” teratoma [8]. The average birth weight of patients with Wilms’ tumors is greater than controls [7].
Symptoms present in less than 10 % of Wilms’ tumor patients as a result of vascular invasion or pressure from surrounding organs. Patients with vascular invasion may present with ascites, congestive heart failure, hepatomegaly, or varicocele. Symptoms caused by tumor-induced hormones include hypertension (25 %), hypercalcemia, erythrocytosis, and von Willebrand’s disease [1, 10, 12].
Ninety percent of Wilms’ tumors are sporadic, and the rest occur as part of overgrowth and non-overgrowth syndromes [8]. Overgrowth syndromes include Beckwith–Wiedemann, isolated hemihypertrophy, Perlman, Sotos’, and the Simpson–Golabi–Behmel syndromes. Non-overgrowth syndromes include WAGR (WT, aniridia, genitourinary anomalies, mental retardation) and Denys–Drash syndromes [1, 10]. Familial cases have an earlier age of onset and an increased incidence of bilateral disease [10].
Imaging Features
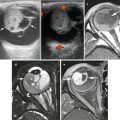
Wilms’ tumor typically appears as a heterogeneous mass arising from the renal parenchyma. The normal renal parenchyma is distorted and displaced around the mass, resulting in a characteristic “claw sign” that helps confirm its renal origin [13]. On sonography, a small WT may appear uniformly isoechoic; however, more often areas of necrosis, hemorrhage, and calcification create a heterogeneous echo texture. On power Doppler evaluation, the mass typically shows decreased perfusion as compared to the normal vascular renal parenchyma. Evaluation of the renal vein with both gray scale and color Doppler evaluation is essential to exclude venous thrombus, which can be present in up to 11.3 % of WT [14]. On CT or MRI, WT typically appear heterogeneous in attenuation/signal intensity and show decreased enhancement as compared to the normal renal parenchyma (Fig. 10.1a). Intracaval extension of the tumor thrombus can be identified with a sensitivity of 84.6–96.0 % on CT (Fig. 10.1b) [15]. The presence and level of tumor thrombus in the IVC determines the surgical approach and need for preoperative chemotherapy, so the renal vein and IVC should be carefully evaluated for any filling defects. Potential pitfalls in the evaluation of the renal vein and IVC include contrast mixing artifacts and extrinsic compression of the vessels caused by a large WT.
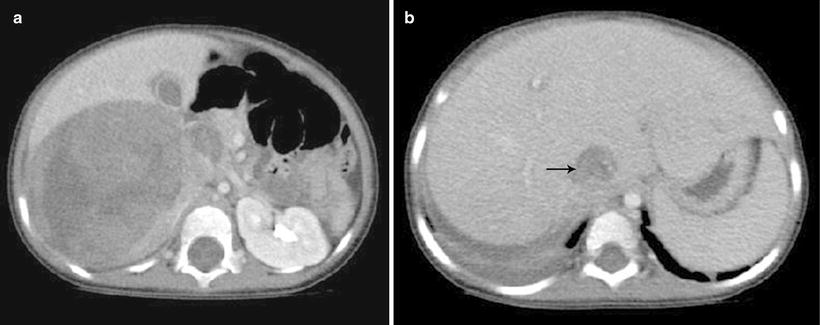
Fig. 10.1
Wilms’ tumor with IVC thrombus. (a and b) Axial contrast enhanced CT images from a 4-year-old show a large heterogeneous mass in the right renal fossa with tumor thrombus distending the inferior vena cava (b) (arrow)
Though microscopic extension of WT through the renal capsule is impossible to detect on imaging, gross infiltration into adjacent structures such as the liver and psoas muscle should be evaluated on imaging. Wilms’ tumor can rupture preoperatively upstaging the child to stage III with increased risk of intraabdominal recurrence. Signs of preoperative tumor rupture include ascites beyond the cul-de-sac, extracapsular perinephric fluid, and peritumoral fat stranding [16]. The regional lymph nodes, lung, and liver, common sites of metastatic disease in WT, should be carefully evaluated. Enlarged lymph nodes should be noted, though current imaging techniques have limited accuracy in detection of nodal metastasis, necessitating sampling during nephrectomy [5]. The contralateral kidney should be carefully evaluated for a contralateral WT or nephrogenic rest, as WT can be bilateral in approximately 10 % of cases [17]. Differentiation of WT from a nephrogenic rest is limited by imaging, though the former tends to inhomogeneous as compared to the homogeneous appearance of rests [18].
Pathology
Gross and Microscopic Features
The majority of Wilms’ tumors form isolated, well-demarcated masses separated from the adjacent kidney (Fig. 10.2) [1, 8]. Rarely, a botryoid variant of Wilms’ tumor is seen, characterized by polypoid mass occupying the renal pelvis (Fig. 10.3). After preoperative chemotherapy, significant amounts of necrosis or hemorrhage can occur. Cystic lesions require careful inspection for nodular solid areas [8].
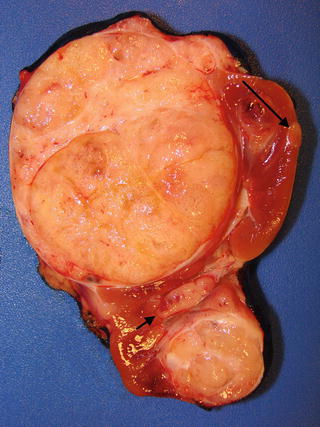
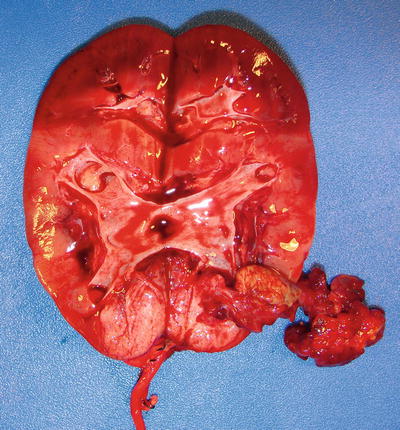
Fig. 10.2
Multifocal Wilms’ tumor with renal sinus invasion (short arrow) and perilobar nephrogenic rest (long arrow)
Fig. 10.3
Botryoid variant of Wilms’ tumor, figure shows tumor arising in the lower pole of the kidney with a polypoid mass attached to the original tumor with a pedicle
Histology is an important prognostic indicator for Wilms’ tumor. Certain histologies, particularly anaplastic Wilms’ tumors, show higher risks of tumor recurrence or chemotherapy resistance [1]. Traditional Wilms’ tumors contain varying amounts of three basic histological components—blastemal, epithelial, and stromal cells—which vary widely in relative proportions (Fig. 10.4) [7, 8]. If one component comprises >2/3 of a tumor, it is referred to as predominant for that specific cell type [7].
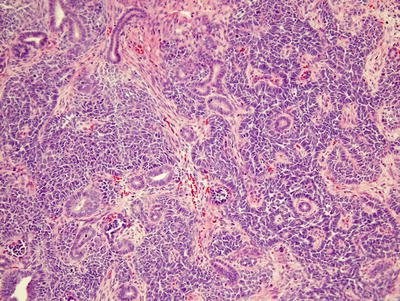
Fig. 10.4
Triphasic Wilms’ showing epithelial, blastemal, and stromal components
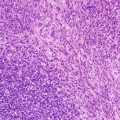
Blastemal predominant Wilms’ tumors are generally more aggressive, i.e., higher stage, but usually respond to stage-specific therapy [10]. They contain sheets of small, round-to-ovoid cells with irregular nuclei, small nucleoli, and little cytoplasm, usually with mitotic figures, apoptotic bodies, and closely packed, overlapping nuclei with diffuse chromatin (Fig. 10.5) [8]. On occasion, rosette formation may resemble neuroblastoma. Growth patterns lack prognostic significance and include diffuse, serpentine, nodular, and basaloid features [7, 8].
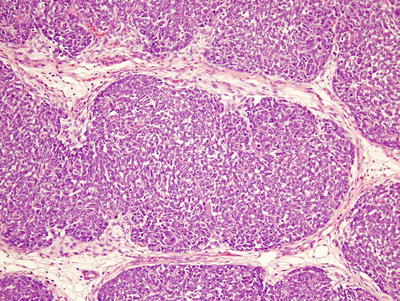
Fig. 10.5
Blastemal predominant Wilms’ tumor. The tumor is composed of primitive small cells arranged in a serpentine pattern and separated by fibrovascular networks
Epithelial predominant Wilms’ tumor is less aggressive but may be resistant to therapy with advanced stage [10]. It is composed of rosette-like and glomeruloid tubules lined by low columnar cells with hyperchromatic nuclei and papillary intratubular invaginations (Fig. 10.6) [7, 8]. Mucinous and squamous epithelium, neural structures, and neuroendocrine cells may also be seen [7]. With epithelial predominant Wilms’ tumor, one should exclude metanephric adenoma (MA), a rare benign tumor that accounts for ~0.2–1 % of all kidney tumors [19]. MAs occur at all ages, ranging 5–80 but most commonly 40–60 years old [8, 20]. Up to 12 % of MA patients have polycythemia [21]. They are purely epithelial lesions lacking stroma and containing densely packed uniform ovoid cells with even, lymphocyte-like nuclei forming a tubular pattern, pink to clear cytoplasm and frequent psammoma bodies. Unlike Wilms’ tumors, MAs contain extremely rare to absent mitoses [21], and show little or no compression of adjacent renal tissue. Metanephric adenofibromas contain both stromal and epithelial components but occur extremely rarely (only 25 cases in the NWTS series [22, 23]).
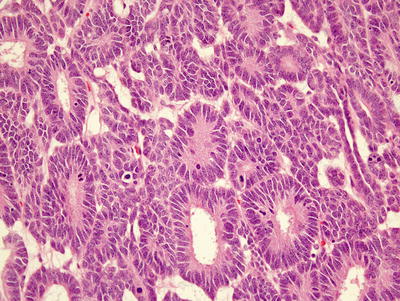
Fig. 10.6
Epithelial predominant Wilms’ composed of rosette-like tubules lined by columnar epithelium with occasional mitotic figures
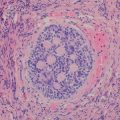
Stromal predominant Wilms’ tumors, like epithelial-predominant ones, show poor clinical responses to chemotherapy [10]. They may contain hypocellular, sparse regions of immature stellate cells in a myxoid background and dense areas of primitive spindled mesenchymal cells. Other stromal cell types include smooth and striated muscle (Fig. 10.7a), adipose tissue, cartilage (Fig. 10.7b), bone, and osteoid, with muscle representing the most common differentiated stromal cell type [7, 8]. It is important in bone forming lesions to consider ossifying renal tumor of infancy, an extremely rare pediatric renal neoplasm with a benign clinical course [23]. With stromal lesions, one should also consider metanephric stromal tumor (MST), an extremely rare, entirely stromal lesion in the spectrum of metanephric neoplasms. Most MSTs occur in the first decade of life [8, 25, 26]. Characteristic concentric “onion skin” rings are observed surrounding entrapped renal tubules or blood vessels. These rings or collarettes are the most defining histological characteristic of MST.
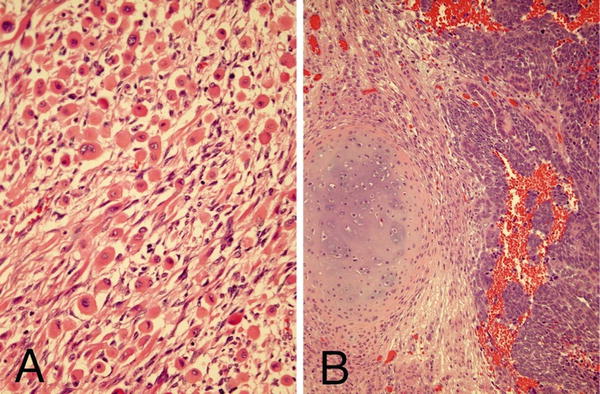
Fig. 10.7
Stromal component of Wilms’ showing skeletal muscle differentiation (a) and cartilaginous differentiation (b)
Anaplasia is the major unfavorable histological variant of Wilms’ tumors and accounts for 5–10 % of unilateral Wilms’ tumors [1, 8, 10]. It rarely occurs in children under 2 but peaks at about 5 years of age [7, 11]. It is more common in African-American that Caucasian patients [11]. Anaplasia is defined by atypical multipolar mitotic figures, hyperchromatism, and enlarged tumor nuclei at least three times the size of adjacent ones (Fig. 10.8). Anaplasia may be focal or diffuse. With focal anaplasia, only one or two isolated intrarenal foci show anaplastic changes. Diffuse anaplasia occurs in extrarenal or multiple intrarenal locations. In stages higher than stage I, diffuse anaplasia has a significantly worse prognosis and is chemotherapy-resistant [7, 8].
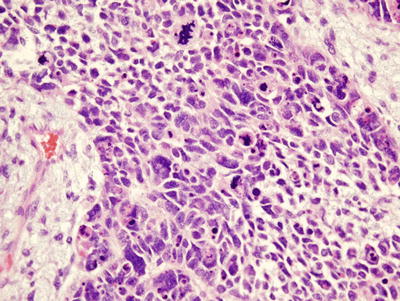
Fig. 10.8
Anaplastic Wilms’ showing a multipolar mitotic figure and large anaplastic cells
Immunohistochemistry and Other Special Stains
Immunohistochemistry has a minor role in diagnosis of nephroblastomas, since the major diagnostic tool lies in tumor morphology. However, it can be helpful with needle core biopsies containing small round cell tumor. WT1, the most helpful marker, is expressed by nuclei of both blastemal and primitive epithelial cells. Primitive renal blastema also shows diffuse and strong but nonspecific positivity for CD56. Nuclear p53 expression has been reported in most anaplastic Wilms’ tumors [8].
Molecular Diagnostic Features and Cytogenetics
Various genes have been implicated in hereditary and syndromic Wilms’ tumor. One to 3 % of patients have a family history of inherited tumors [1, 9]. Genetically inherited cases tend to have an earlier onset and increased bilaterality [10]. Two familial genes have been specified: FWT1 at 17q12-q21 and FWT2 at 19q13 [10]. About 10 % of Wilms’ tumor patients show associated congenital abnormalities and syndromes. Mutations of Wilms’ tumor 1 (WT1), a tumor suppressor gene located at 11p13, occur in the Wilms’ tumor, aniridia, genitourinary anomalies, mental retardation (WAGR), and Denys–Drash (DDS) syndromes (Fig. 10.9) [9]. WT1 encodes a transcription factor important in gonadal development, ureteric budding, and nephrogenesis [1]. WAGR and DDS patients have increased risk of bilateral tumors, younger age, and renal dysfunction [10]. WT1 mutations also occur in about 2 % of sporadic tumors [10].
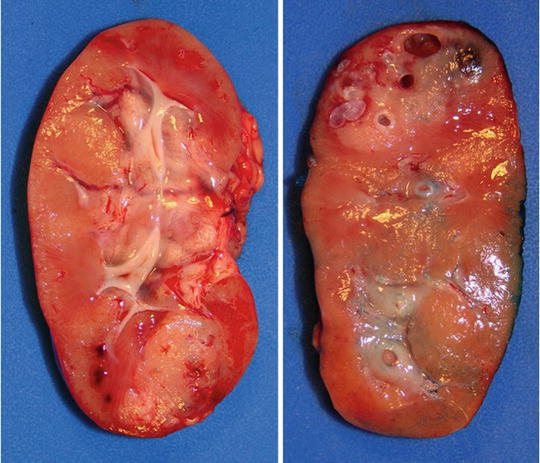
Fig. 10.9
Bilateral Wilms’, nephrectomy in a patient with Denys–Drash syndrome
Aniridia is a non-overgrowth syndrome found in 1.1 % of Wilms’ tumor patients. It is caused by a defect in the PAX6 gene located at 11p13 adjacent to WT1. About 40–70 % of aniridia patients have a contiguous WT1 deletion causing Wilms’ tumor [1, 10]. In addition, more than half of tumors with WT1 mutations have coexisting ones in the beta-catenin gene (CTNNB1), suggesting involvement of the Wnt/beta-catenin pathway, and mutations in this gene are found in 5–15 % of Wilms’ tumors overall [9, 10].
Loss of heterozygosity (LOH) of the Wilms’ tumor 2 (WT2) gene locus located on chromosome 11p15 has been identified in up to 40 % of sporadic cases and 1–8 % of Beckwith–Wiedemann syndrome (BWS) patients [9]. BWS patients with hemihypertrophy have a 4–10 % chance of developing Wilms’ tumors, 21 % of them bilateral [1, 10]. Specific genes located within the WT2 domain include H19 and insulin-like growth factor 2 (IGF2) [10]. Differential DNA methylation of these two genes reveals biallelic expression of IGF2 in 26–77 % and hypermethylation of H19 in 26–75 % of Wilms’ tumors without LOH [9].
Wilms’ tumors show LOH at chromosomes 16q and 1p in 20 % and 10 % of cases, respectively. LOH at these locations increases the risk of tumor relapse and mortality [1, 10]. Lesions that have LOH at both 16q and 1p show an even greater propensity for relapse and mortality, with the relapse-free rate dropping to 74.9 % (in combination) from 80.4 % (1p alone) and 82.5 % (16q alone) [27]. LOH at 11q occurs in 20 % of Wilms’ tumors and is three to four times more common in anaplastic ones [10].
Prognostic Features
Although survival for patients diagnosed with Wilms’ tumor was once under 30 %, more than 90 % of children now have excellent outcomes [1, 11]. This marked improvement is due to adherence to protocol-based therapy that includes specific surgical approaches, enhancements in chemotherapy, and refined radiation therapy [11].
Histopathology is a major prognostic indicator, especially after preoperative chemotherapy. Blastemal Wilms’ tumors are highly aggressive, and more than 75 % of this subtype present at stage III or IV. Blastema is generally responsive to chemotherapy but is associated with a high relapse rate [7, 10]. Substantial reduction in size subsequent to chemotherapy is a good prognostic indicator [7], but persistent viability after chemotherapy of a blastemal lesion indicates a poor prognosis [8]. Diffusely anaplastic tumors are resistant to chemotherapy but with complete excision have better prognosis [1]. Epithelial predominant tumors are less aggressive, and more than 80 % present at stage I. Although more resistant to chemotherapy, epithelial and stromal-predominant subtypes have a good prognosis after complete resection [1, 7].
Wilms’ tumor metastasizes most commonly to the lungs and less commonly to the liver. About 12 % of patients show hematogenous metastases at diagnosis, 80 % extending to the lungs. Lymph node involvement indicates distant metastases but is difficult to assess [1, 7, 8, 11]. About 20 % of favorable histology tumors relapse after therapy [11], but patients are usually curable if there is no liver or mediastinal metastasis. However, repeated relapses indicate a very poor prognosis [7].
LOH for chromosomes 1p and 16q in stage I and II Wilms’ tumors confers a significantly worse prognosis and is now used to stratify Wilms’ tumor patients into different levels of risk and treatment [11].
Nephrogenic Rests and Nephroblastomatosis
Nephrogenic rests (NRs), abnormally persistent clusters of embryonal renal parenchyma tissue beyond 36 weeks of gestation, are known precursors of Wilms’ tumors (WT) [28–30]. Multifocal or diffuse lesions are called nephroblastomatosis. Based on their topographic location with respect to the renal lobe, nephrogenic rests are separated into perilobar nephrogenic rests (PLNR), found at the periphery of the lobe (often subcapsular), and intralobar nephrogenic rests (ILNR), present anywhere within the kidney [8, 31, 32]. Both of these subtypes can be further classified as dormant, sclerosing, hyperplastic, or neoplastic (development into WT), depending on their eventual fate [29–32].
Nephrogenic rests are often clinically asymptomatic and are usually discovered incidentally in conjunction with Wilms’ tumor [30]. Higher than average birth weight significantly correlates with NR diagnosis, especially PLNR. A small percentage of patients with NRs have associated syndromes or genetic abnormalities. WAGR and Denys–Drash syndromes are strongly associated with ILNR, along with other genital anomalies like hypospadias and cryptorchidism. Overgrowth syndromes like BWS and hemihypertrophy (HH) have a higher prevalence of PLNR but a slight correlation with ILNR. PLNRs also occur in Perlman syndrome, and trisomies 13 and 18 [29, 33].
The subtypes and various fates of NRs can be histologically distinguished based on their location, gross features, and morphologic appearance. Perilobar nephrogenic rests are predominantly composed of well circumscribed blastemal tissue (Fig. 10.2; long arrow), while ILNRs are primarily composed of stromal or epithelial tissue with irregular margins that often interdigitate with surrounding normal interstitial tissue. PLNRs usually present with multiple lesions that well demarcated from adjacent kidney, while ILNRs mostly occur as isolated or few foci that often blend with adjacent kidney [7, 30].
The pathogenesis of NRs is undoubtedly linked to that of Wilms’ tumor, with perhaps over ten genes involved. Many studies have reported similar LOH in both NRs and adjacent Wilms’ tumors [34–37]. Since PLNR is positioned in the periphery of the renal lobe, their genetic dysregulation likely occurs later in development than ILNR, following lobar pattern establishment. The deeper location of ILNR within the renal lobe supports an earlier developmental abnormality [29].
Imaging Features of Nephrogenic Rests
Recent advancements in imaging technology have greatly facilitated the detection of small renal tumors and nephrogenic rests. Both contrast enhanced CT and contrast enhanced MRI can detect nephrogenic rests as small as 4–5 mm, so routine intraoperative exploration of the contralateral kidney is no longer recommended [38].
Because US is widely available and relatively inexpensive, it is the primary imaging method for screening patients at risk of nephrogenic rests. On US, NR lesions are typically homogeneous and hypoechoic or isoechoic to normal kidney tissue. As nephrogenic rests are relatively less vascular than normal renal parenchyma, use of power Doppler imaging can aid in the detection of nephrogenic rests. Ultrasonography can detect NR as small as 8 mm, but has a lower sensitivity than CT or MRI [18, 39, 40]. On CT, nephrogenic rests are most evident on post contrast images as homogeneous lesions that enhance less than the normal renal parenchyma. On MR, hyperplastic rests are relatively T2 hyperintense compared to normal renal parenchyma, while sclerotic rests are relatively hypointense on T2 weighted images [39]. As on CT, nephrogenic rests enhance less than normal renal parenchyma on post contrast MR images.
Imaging differentiation of nephrogenic rests from Wilms’ tumor can be difficult. While nephrogenic rests tend to have a more homogeneous appearance on all imaging modalities, heterogeneity favors Wilms’ tumor. Both tumor and hyperplastic rests tend to be bright on T2-weighted images [39]. The size of the lesion per se is not reliable for differentiation, as nephrogenic rests up to 5 cm in diameter have been reported [18].
The presence of multiple bilateral nephrogenic rests is referred to as nephroblastomatosis (Fig. 10.10a, b) [41]. A specific subtype of nephroblastomatosis is diffuse hyperplastic perilobar nephroblastomatosis (DHPLN), characterized by a rind of nephroblastic tissue surrounding the renal parenchyma (Fig. 10.11). On imaging the kidneys are diffusely enlarged but maintain their normal reniform shape. The nephroblastic rind is confined to the periphery of the kidney, has homogeneous attenuation/signal intensity, and enhances less than the normal kidney.
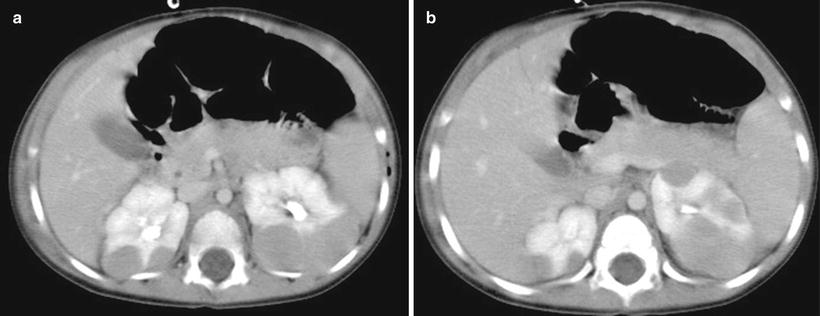
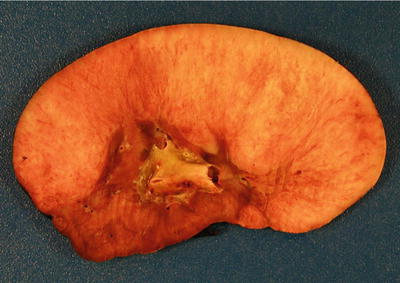
Fig. 10.10
Bilateral Wilms’ tumor (nephroblastomatosis). (a and b) Axial contrast enhanced CT images of the abdomen in a 3-year-old show bilateral hypodense lesions consistent with nephroblastomatosis
Fig. 10.11
Diffuse nephroblastomatosis showing overgrowth of perilobar nephroblastic tissue
Nephroblastomatosis should be distinguished from renal lymphoma, which can also result in bilateral renal masses. Children with nephroblastomatosis are younger (<5 years of age) and lack the extensive retroperitoneal lymphadenopathy usually seen in patients with lymphomatous involvement of the kidneys.
Cystic Nephroma and Cystic Partially Differentiated Nephroblastoma
Cystic nephroma (CN) and cystic partially differentiated nephroblastoma (CPDN) are two uncommon benign renal tumors macroscopically characterized by multilocular cysts. The two entities are well demarcated from the rest of the kidney and have no solid components except for septa composed of fibrous tissue. A distinction is drawn between CN and CPDN in that the former contains only mature septal elements such as tubules while the latter contains embryonal elements or blastema in the cyst walls [7, 8].
For CN and CPDN, there exist four criteria that characterize the lesion. First, the entity is composed entirely of cysts and their septa. The lesion is discrete from the surrounding renal parenchyma and well demarcated. The septa are the only solid component of the tumor and completely encase the cysts without any solid expanding nodules. Finally, the cysts are lined by flattened, cuboidal, or hobnail epithelium [42].
Cystic nephroma is distinguished by its fibrous septa that is well differentiated and contains only mature elements such as tubules. Also, mature tissue such as heterologous skeletal muscle can also be seen in CN.
Cystic partially differentiated nephroblastoma macroscopically and microscopically resembles CN but contains various immature septal elements. Usually this difference is characterized by poorly differentiated blastemal cells in the septa that may contain other embryonal cell types such as stroma or epithelia comprising poorly differentiated glomeruli, cartilage, fibrous tissue, mesenchyme, fat, tubules, or striated muscle intermixed with blastemal cells [42].
CPDN shows no excessive mitotic activity or evidence of contiguous extension or vascular involvement. The “partially differentiated” term is derived from the mixture of undifferentiated blastemal tissue with partially or well-differentiated renal tissue [43]. Cystic nephroma does not present with this spectrum of differentiation.
CN is benign,as tumor-related death or metastasis has not been previously reported. Local recurrence of the lesion after resection has been reported, most probably due to outgrowth from residual tissue and not malignancy [44].
Cystic partially differentiated nephroblastomas have an equally favorable prognosis with 100 % survival in some studies. This applies to stage 1 and 2 CPDN, which are both successfully treated with complete resection of the tumor and varying amounts of chemotherapy [45]. However, one study has reported two cases in which CPDNs undertook a more aggressive course and lead to death [43]. The more aggressive potential of CPDN may be due to the malignant potential of the poorly differentiated tissue present in the septa. The extremely favorable prognosis of CN and CPDN provides additional separation from Wilms’ tumor, which has a less favorable prognosis.
Imaging Features of Cystic Tumors
Cystic nephroma and cystic partially differentiated nephroblastoma are relatively uncommon benign lesions that are indistinguishable on imaging [46]. They are typically solitary, multilocular lesions sharply demarcated from an otherwise normal remaining kidney. The masses are usually large, averaging 8–10 cm in size, and involve only part of the kidney. These are multicystic lesions, and with no solid nodules, that may protrude into the renal pelvis/ureter. On sonography, the mass consists of multiple anechoic cystic areas separated by septations (Fig. 10.12a, b). These cystic areas can vary in size from microscopic to 4 cm in diameter. The microcystic areas may mimic solid areas due to the closely packed septations. On CT and MRI, CN and CPDN typically appear as well circumscribed, multicystic masses that have variable enhancement of septations on the post contrast images. The septations are typically hypointense on precontrast T1 and T2 weighted images due to fibrous tissue in them. The differential diagnosis of a multilocular cystic renal mass in a child includes cystic Wilms’ tumor or renal cell carcinoma; clear cell sarcoma; cystic variants of mesoblastic nephroma; and segmental forms of multicystic dysplastic kidney. The presence of solid components within a cystic mass should make one consider a diagnosis other cystic nephroma/cystic partially differentiated nephroblastoma. If a cystic pulmonary lesion is identified in a child with a multicystic renal mass, this should lead one to consider a DICER 1 mutation with cystic nephroma and coexistent pleuropulmonary blastoma [47].
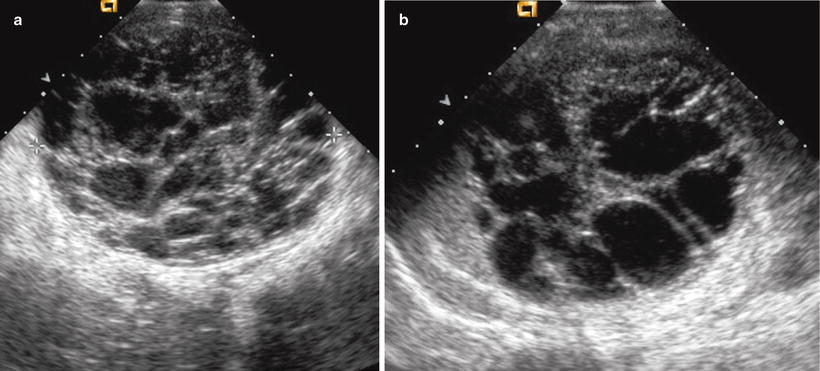
Fig. 10.12
Multilocular cystic nephroma. (a and b) Sonographic images from a 9 month old show a multiloculated cystic mass arising from the left kidney. No solid components are noted. Pathology confirmed a multilocular cystic nephroma
Mesoblastic Nephroma
Definition
Clinical Features and Epidemiology
Incidence and Prevalence
Although CMN only represents less than 5 % of all pediatric renal neoplasms, it is the most common congenital renal tumor, with over 90 % of patients diagnosed before age 1 and almost no patients presenting after the age of 3. The cellular variant represents approximately 40–60 % of CMN, with the classical and mixed subtypes constituting the remaining percentage with about an equal prevalence [8, 48].
Population Features
The majority of renal tumors diagnosed within the first 6 months of life are CMN, so it must be included in the differential diagnosis of any infant with a renal mass. Because its incidence drops off significantly after early childhood, diagnosis of any spindle cell tumor as CMN must be questioned in patients older than 3 years. There does not appear to be any definitive gender predisposition, but some studies report a slight male prevalence [49, 50]. The general median age of diagnosis is approximately 20–35 days, with the cellular type (median: 3–4 months) being diagnosed at a significantly older age than the classic (median: 7 days) or mixed (median: 2 months) variants [49, 50].
Presenting Symptoms and Signs
Most patients present with an asymptomatic abdominal mass discovered upon physical examination or after imaging analysis. Other symptoms include abdominal swelling and protrusion, abdominal pain, hypertension, hypercalcemia, and hematuria [48, 50, 51]. These tumors may cause polyhydramnios during pregnancy, requiring preterm delivery in a reported 71 % of CMN associated fetuses [48].
Imaging Features
Mesoblastic nephroma presents as a solitary unilateral mass with variable cystic/solid mass in the neonate, typically within the first 3 months of life [52]. Mesoblastic nephroma has been detected in utero on prenatal sonography as a heterogeneous renal mass as early as 26 weeks. These masses can be associated with polyhydramnios and premature labor [53]. The classic and cellular subtypes of CMN differ in their imaging appearances. Classic CMN presents as a uniform soft tissue mass with minimal, predominantly peripheral, enhancement seen on post contrast CT or MRI. However, the cellular variant of CMN can have a more heterogeneous appearance and can vary from a predominantly cystic to mixed solid and cystic mass (Fig. 10.13a, b). Areas of cystic change, necrosis, and hemorrhage can be seen on all imaging modalities in cellular CMN, corresponding to its pathologic appearance.
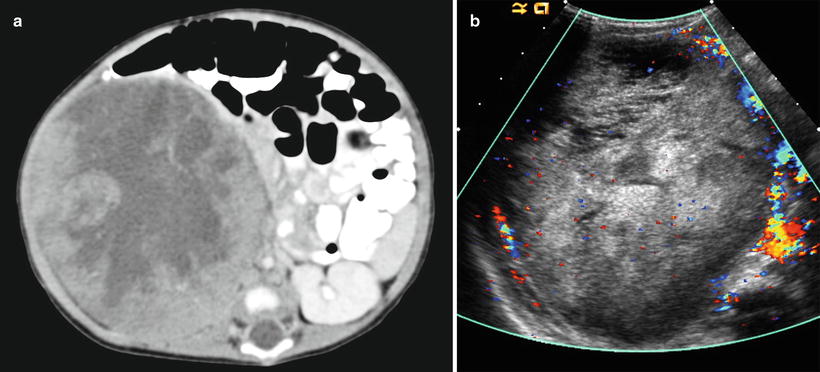
Fig. 10.13
Mesoblastic nephroma. Axial contrast enhanced CT (a) and color Doppler image (b) from a 54 day old neonate shows a heterogeneous, well circumscribed mass consistent with a cellular variant of mesoblastic nephroma
Pathology
Gross and Microscopic Features
Grossly, the tumor is usually solid but rarely can have a prominent cystic appearance. Congenital mesoblastic nephroma tend to be firm pale to tan-colored lesions (Fig. 10.14), with the cellular variant being softer and more likely to demonstrate necrosis or hemorrhage than the classic variant. The mixed subtype shows both characteristics. The tumor often infiltrates normal adjacent renal parenchyma and perinephric fat. As previously mentioned, the cellular type is usually much larger in volume than the mixed or classic types [7, 8, 48].
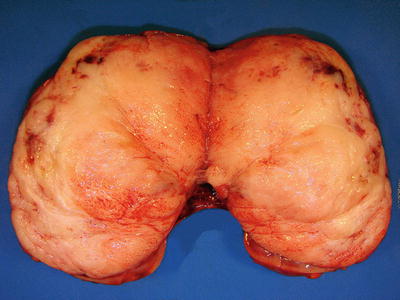
Fig. 10.14
Congenital mesoblastic nephroma showing firm, pale-colored cut surface with small cystic and hemorrhagic areas
Histologically, classic CMN demonstrates morphology similar to that of uterine leiomyoma and infantile fibromatosis; it contains bundled spindle cells, infrequent mitosis, and absence of necrosis (Fig. 10.15a). The spindle cells lie within collagenous stroma, show an interlacing fascicular pattern, and infiltrate surrounding renal parenchyma as finger-like projections. The cellular variant has a more malignant appearance, with an increased nuclear: cytoplasmic ratio, cellularity, and mitotic index and significantly more necrosis and hemorrhage (Fig. 10.15b). CMNs lack the blastemal components observed in Wilms’ tumor and do not demonstrate characteristic vascular patterns, nuclear grooves, or clear cells as seen in clear cell sarcoma of the kidney [8, 51, 52]. However, they often entrap renal tubular elements, which may show atypia.
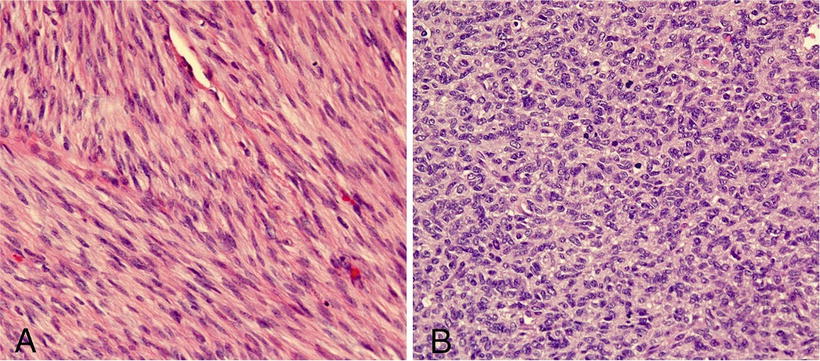
Fig. 10.15
(a) Classic CMN showing interlacing fascicular pattern of benign spindle cells. (b) Cellular variant showing sheets of round cells with increased nuclear cytoplasmic ratio and abundant mitotic figures
Immunohistochemistry and Other Special Stains
The most common immunohistochemical characteristic of CMN is diffuse nonspecific reactivity for vimentin and focal reactivity for smooth muscle actin [8, 51]. The tumor usually does not show immunoreactivity for desmin, S-100, cytokeratin, AE1/AE3, epithelial membrane antigen, bcl-2, or CD99 [51, 54].
Molecular Diagnostic Features and Cytogenetics
Recent cytogenetic studies have found strong genetic correlations between the cellular variant of CMN and congenital infantile fibrosarcoma (CFS). Using RT-PCR and fluorescence in situ hybridization, a study found that five out of six cellular CMN tumors and five out of five CFS tumors tested were found with the same t(12;15)(p13;q25) translocation that resulted in a ETV6-NTRK3 (aka TEL-NTRK3) gene fusion [55]. Both of these entities have also been associated with genetic polysomies at chromosomes 8, 11, 17, and 20 [55, 56], with trisomy 11 being strongly correlated with CMN. Because cellular CMN and CFS have similar histological and morphologic features and the same genetic abnormalities, these two neoplasms appear to represent the same entity, with cellular CMN being the renal variant. Additionally, it has been reported that primary bronchopulmonary fibrosarcoma (BPFS), a rare lower respiratory neoplasm affecting children and young adults, also carries the t(12;15) ETV6-NTRK3 genetic abnormality [55, 57]. Thus, we consider CMN and BPFS as visceral variants of CFS. The same t(12;15) ETV6-NTRK3 fusion does not appear with classic CMN, which may be found in conjunction with cellular CMN in the mixed subtype. ETV6-NTRK3 fusion is also found in a dissimilar tumor, secretory carcinoma of the breast [55].
Prognostic Features
Mesoblastic nephroma is usually a benign neoplasm with overall good prognosis after nephrectomy. Local recurrences and metastasis to the brain, lungs, heart, bone, and liver have been reported, with the majority of these relapses being associated with the cellular variant of CMN. Overall, about 10 % of CMN tumors relapse and such recurrences almost always occur during the first year after initial diagnosis [48]. The survival overall is very high at >95 %. Late stage cellular CMN occurring in patients older than 3 years carries the worst prognosis. Adjuvant chemotherapy is not recommended after complete resection [7, 49].
Clear Cell Sarcoma of the Kidney
Definition
Clear cell sarcoma of the kidney (CCSK) is rare renal pediatric neoplasm with a generally unfavorable prognosis [58, 59]. Because of its greater tendency towards bone metastasis, the tumor has been called bone metastasizing renal tumor of childhood, and it has also been referred to as sarcomatous Wilms’ tumor [7].
Clinical Features and Epidemiology
CCSK is extremely rare; only 20 new cases are reported annually in the USA [60]. Overall accounts for 3–5 % of all pediatric renal neoplasms. Nevertheless, CCSK still represents the second most common childhood renal tumor [61].
CCSK patients present at a mean age of 36 months; most are diagnosed between ages 2 and 3 years, with a large decline in incidence afterwards [60]. It is extremely uncommon during the first 6 months of life and has seldom been reported in adolescents and adults [59, 60]. Patients have a male predominance with an average ratio of 2:1 [7, 59, 60].
Clinical manifestations of CCSK include an abdominal mass or swelling, abdominal pain, constipation, decreased appetite, fever, gross hematuria, hypertension, and vomiting [59, 63]. Approximately 5 % present with metastasis, with the lymph nodes being the most common site [8, 60]. According to the National Wilms’ Tumor Study (NWTS), approximately 26 % of CCSK cases present at stage I, 35 % at stage II, 34 % at stage III, and 5 % at stage IV, with negligible numbers at stage V [59, 60].
Imaging Features
Clear cell sarcoma typically presents as a large, solid but inhomogeneous renal mass with cystic/necrotic areas, that on imaging is indistinguishable from WT [64]. Foci of calcification can be present in up to 25 % of cases. This aggressive tumor can have extracapsular spread and vascular extension at the time of initial presentation, though the latter is less common than in WT cases. The most common sites of metastasis are bone, lymph nodes, lung, and liver.
Pathology
Gross and Microscopic Features
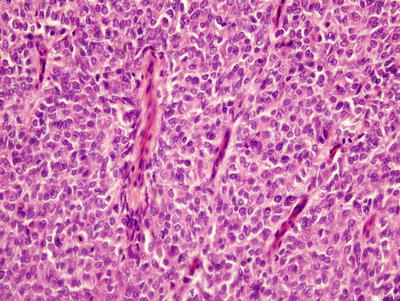
Grossly, CCSKs are large, soft, and gray-tan to white in appearance. They are uniform, fleshy, well circumscribed, sharply demarcated, and almost always present unilaterally in the kidneys. Tumors are usually solid but may be focally cystic with necrosis and hemorrhage [7, 8, 54, 63]. The tumors often involve renal medulla and may replace and distort the entire kidney [7, 59]. Renal vein invasion occurs in approximately 5 % of cases [59, 60]. Tumors measure between 2.3 and 24 cm [60].
Perhaps the most diagnostically difficult aspect of CCSK is its histological variability: nine variations have been described [59, 60]. The classic pattern contains cords or nests of ovoid, epithelioid, and spindle shaped cells separated by consistently spaced fibrovascular septa [67, 60]. These septa characteristically feature branching or “chicken-wire” capillaries that vary in width. Tumor nuclei are uniform in shape, and their cytoplasm is clear to pale with an ill-defined cell border (Fig. 10.16) [60, 63]. Although over 90 % of CCSK predominately or focally demonstrate the classic pattern, most also present one of the other eight identified histologic variants, percentages: myxoid (50 %), sclerosing (35 %), cellular (26 %), epithelioid (13 %), palisading (11 %), spindle cell (7 %), storiform (4 %), and anaplastic (2.6 %) [60]. The different patterns are essentially varying alterations of the cord/nest or septal cell appearance, and they therefore cannot be separated as distinct biological entities.