Fig. 13.1
Multiple hyperpigmented macules and a basal cell carcinoma in a patient with xeroderma pigmentosum (Courtesy of Dr. A. Torrelo, Madrid)
Patients with XP under 20 years of age have a greater than 1,000-fold increased risk of developing skin cancer (BCC, SCC, or MM) [7]; multiple, simultaneous primary skin cancers are common in these patients. The median age of onset of NMSC reported in patients with XP is 8 years. MM develops in about 5 % of XP patients [9], with an estimated median age of onset of 19 years [7], although one study reports that up to 40 % of XP-associated melanomas occur in children under 12 years of age [10]. Children affected by XP must be followed very closely and observe extreme sun protection.
Familial Atypical Mole Melanoma Syndrome
Familial atypical mole melanoma syndrome (FAMMS), sometimes called dysplastic nevus syndrome, or familial cutaneous melanoma syndrome, is characterized by familial MM in the setting of atypical moles. Components of the syndrome include a family history of cutaneous MM in two or more first-degree relatives, early age of diagnosis, and the presence of numerous (usually more than 100) often clinically atypical melanocytic nevi, and these are usually 5–10 mm in diameter, variegated from tan to dark brown, and round or oval with irregular, ill-defined borders. They often feature a central elevation within a flat lesion. Early in childhood, the nevi may appear relatively normal, but the atypical nevus phenotype is fully apparent at puberty in most patients [1].
The genetic susceptibility to develop MM in these patients is caused by germ line mutations in the CDKN2A, but rare cases of families carrying mutations in the CDK4 gene have also been described. Through alternative splicing, CDKN2A encodes two distinct protein products, p16 and p14ARF, that exert inhibitory effects on cell growth through distinct pathways involving the retinoblastoma and p53 proteins [11]. The prevalence of germ line CDKN2A mutations among familial melanoma kindreds varies widely, ranging from 20 % in Australia, to 45 % in North America and 57 % in Europe [12].
Nevoid Basal Cell Carcinoma Syndrome
The nevoid basal cell carcinoma syndrome (NBCCS), or Gorlin syndrome, is an autosomal dominant genodermatosis resulting from heterozygous mutations in PTCH1, PTCH2, SUFU, or other as yet unknown genes [15–18]. NBCCS is transmitted as an autosomal dominant trait with complete penetrance and variable expressivity [17].
NBCCS is characterized by numerous BCC and epidermal cysts of skin, odontogenic keratocysts in the jaws, palmar and plantar pits, calcified dural folds, various neoplasms or hamartomas (ovarian fibromas, medulloblastoma, lymphomesenteric cysts, fetal rhabdomyomas, etc.), and various stigmata of maldevelopment (rib and vertebral abnormalities, Sprengel anomaly, enlarged head circumference, cleft lip and/or palate, cortical defects of bones, etc.) (Fig. 13.2). Diagnosis of NBBCC requires two major or one major and two minor clinical criteria (Table 13.1) [19]. BCCs may arise at various stages of the syndrome; most often they appear between puberty and 35 years of age [20], but patients as young as 4 years old have been reported [21]. Different histological subtypes of BCC, even affecting the same patient, have been described [22]. Documented histological patterns include infundibulocystic, nodular, superficial, pigmented, and many others [23]. The appearance of odontogenic keratocysts and new BCCs continues throughout life. Medulloblastomas appear before the age of 4 years, while ovarian fibromas usually develop after puberty. A rare case with the simultaneous occurrence of two solid tumors commonly seen in the pediatric age, rhabdomyosarcoma and Wilms tumor, has also been described [24]. Therapeutic radiation should be avoided whenever possible due to the high occurrence of BCC in the radiation field. Limitation of sun exposure reduces the appearance of skin cancers.
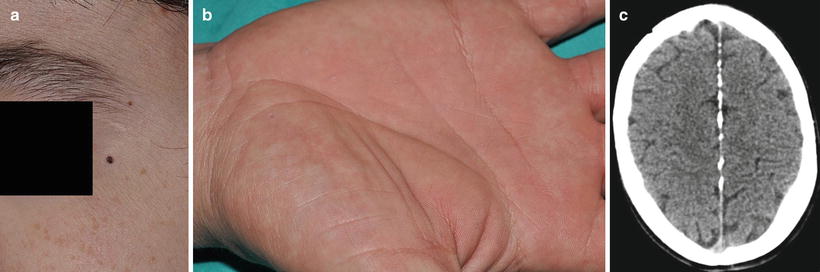
Fig. 13.2
Clinical features of the nevoid basal cell nevus syndrome. (a) Pigmented basal cell carcinoma, (b) palmar pits, and (c) falx cerebri calcification (Courtesy of Dr. A. Torrelo, Madrid)
Major criteria |
1. More than two BCCs or one under age of 20 years 2. Odontogenic keratocysts 3. Three or more palmar pits 4. Bilamellar calcification of falx cerebri 5. Bifid, fused, or splayed ribs 6. First-degree relative with NBCCS |
Minor criteria |
1. Macrocephaly adjusted for height 2. Frontal bossing, cleft lip/palate, hypertelorism 3. Sprengel deformity, pectus, syndactyly of digits 4. Bridging of sella turcica, hemivertebrae, flame-shaped radiolucencies 5. Ovarian fibroma 6. Medulloblastoma |
Two major or one major and two minor criteria are necessary to diagnose NBCCS |
Malignant Melanoma
Pediatric MM is generally defined as melanoma occurring in patients ranging in age from in utero to 21 years, although the upper limits of the cutoff age vary from 13 to 21 years in published reports. Pediatric melanoma can be subdivided into several groups including congenital (in utero to birth), neonatal or infantile (birth to 1 year), childhood (1 year to puberty), and adolescent melanoma (puberty to 21) [25]. In children, as in adults, most melanomas develop de novo. In children, atypical, amelanotic, and nodular melanomas are more common [26].
Epidemiology
MM is the most common skin cancer of childhood, followed by BCC and SCC. It accounts for <3 % of all cancers seen in children, and the incidence in children and adolescents accounts for only 1 % of all new cases diagnosed in the United States annually. The incidence of pediatric melanoma in the United States appears to have increased from 1973 to 2001 at a rate of 2.9 % per year and 46 % per year of age [27, 28]. The incidence of pediatric MM is higher in older children. A review of the National Cancer Institute Registry for melanoma patients aged 1–19 found that 3.8 % were between 1 and 4 years old, 5.7 % were 5 and 9 years old, 17.3 % were 10 and 14 years old, and 73 % were 15 and 19 years old [27–29].
Whites are more likely to develop MM than any other racial-ethnic group. However, there is a proportionately higher incidence of melanoma in nonwhite children less than 10 years of age [27]. There is an apparent female predominance, and the female-to-male ratio increases with age during adolescence and young adulthood. Melanoma can present at any site; however, the extremity is the most common primary site of disease in patients under 20 years of age, followed by trunk and head and neck [30].
Pathogenesis and Genetics
The exact etiology of MM in children remains unclear. It is likely that there is interplay of both inherited and environmental factors.
MM development has been linked to germ line mutations in genes encoding CDKN2A, CDK4, and MCIR as well as to somatic mutations in proto-oncogenes B-RAF, N-RAS, KIT and tumor suppressor genes CDKN2A, p53, and PTEN. Somatic mutations in B-RAF are the most common genetic alterations in MM, occurring in up to 66 % of cases. There are at least 30 documented mutations of B-RAF that activate the MAPK pathway. The most common B-RAF mutation is V600E [31]. Targeting these mutations with specific inhibitors offers an exciting new therapeutic approach for these tumors.
Uribe et al. [32] recently described a pediatric-specific pathogenetic finding. Higher levels of microsatellite instability (MSI) and LOH were found in pediatric melanoma as compared with adult melanoma, although the differences did not reach statistical significance. Increased allelic loss at 11q23 was found in pediatric melanoma, postulated to be related to its early onset. The higher MSI found in pediatric MM could increase the rate of spontaneous mutations in both oncogenes and tumor suppressor genes, leading to tumorigenesis. Increased loss of heterozygosity (LOH) involving TP53, RB1, and BRCA1 in both adult and pediatric melanoma possibly reflects inactivation of these genes and a role in melanoma pathogenesis [25].
Risk Factors
Most of the predisposing factors shown in Table 13.2 lead to melanomas in adulthood, although they may have been present since childhood [32]. In addition to the already mentioned inherited tumor syndromes such as XP and FAMMS, significant predisposing conditions include congenital melanocytic nevi, immune dysfunction (either immunodeficiency or immunosuppression), and a previous history of another malignancy [27, 29, 33]. Immunosuppression secondary to a hematologic, infectious, or acquired disorder (e.g., organ or bone marrow transplant) increases the pediatric risk of melanoma by three- to sixfold [34].
Table 13.2
Predisposing factors for malignant melanocytic skin tumors in children
• Impaired DNA repair, especially xeroderma pigmentosum (XP) |
• Familial atypical mole syndrome (FAMM) |
• Immunosuppression |
• Previous malignant disease |
• Congenital melanocytic nevus, especially giant nevus |
• Melanoma in the family |
• Large number of common melanocytic nevi |
• Several atypical melanocytic nevi |
• Light skin, red or blond hair, and/or light eyes |
• Freckles and/or actinic lentigines (sun spots) |
• Tendency to sunburn when exposed to UV light |
• Intermittent intensive exposure to UV light |
Congenital Melanocytic Nevi (CMN)
Figure 13.3 represent benign proliferations of melanocytes that present at birth or within the first few months of life [35, 36]. Krengel et al. [37] have proposed a system that offers an objective and reproducible classification.
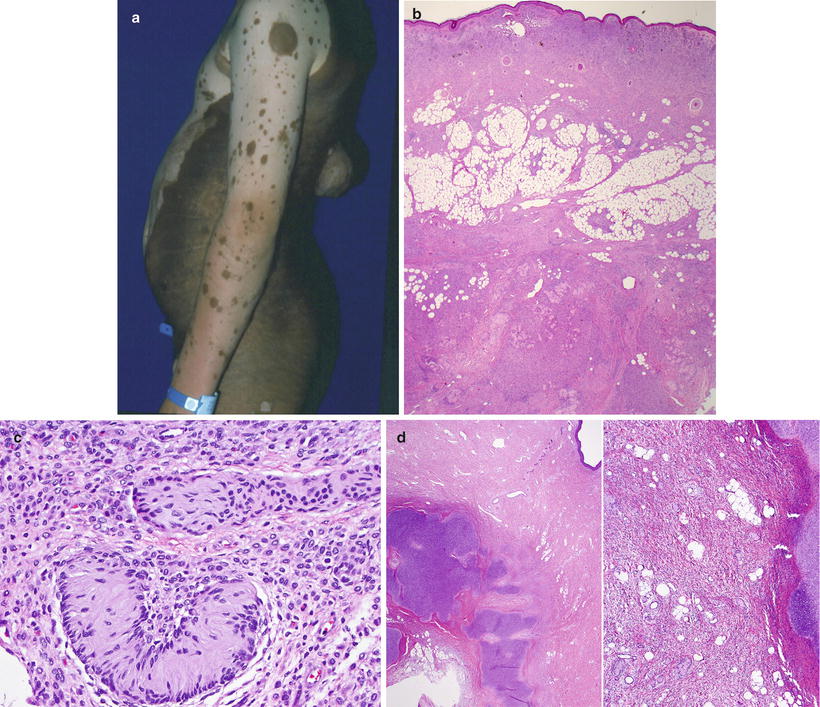
Fig. 13.3
Giant congenital melanocytic nevus of the “bathing-trunk” type with multiple “satellite” nevi and a nodular malignant melanoma in the back (a). On low power, the nevus cells replace the entire dermal compartments and extend through the subcutaneous tissue and underlying fascia (b). The so-called neurotization is represented by the formation of Wagner-Meissner bodies, or lames foliacées (c). Hyaline cartilage is a common heterologous component seen within giant congenital nevi (d)
Giant CMN are larger than 20 cm in final size. In neonates, giant CMN can be alternatively defined as being larger than the palm of the patient at birth [38]. Giant CMNs are rare and occur in approximately one in 20,000 births [39]. They cause significant distress for the patients and their families, due to their marked cosmetic effects and purported malignant potential. Giant CMN are most commonly found on the posterior trunk (“bathing trunk nevi”) and are characterized by a verrucous surface, significant pigmentation, color variation within the dominant background color, and irregular margins. They are accompanied by smaller “satellite” nevi (Fig. 13.3a). As the infant grows, the lesion often acquires increased terminal hairs, variegated colors, and a more irregular surface. Changes may also occur at puberty [40]. Giant congenital nevi have a peculiar histological appearance (vide infra) and frequently show an extensive and deep involvement of the entire dermis and subcutaneous tissue, sometimes going even deeper into fascial planes. They may feature significant “neurotization,” represented by formations that recapitulate the neural crest origin of these cells, with Masson or Wagner-Meissner bodies, also known as lames folliacées. Heterologous elements may also be found, probably indicative of the multipotential differentiation capabilities of the neural crest elements from which they form (Fig. 13.3b–d). In addition, deep tissues underlying these congenital nevi frequently feature poorly developed muscle and vascular elements, resulting in decreased or altered strength and other local symptoms (MRM, unpublished observations). Medium-size congenital nevi are defined as those >1.5 cm but <20 cm in diameter. They occur in about 0.6 % of newborns. Small congenital nevi are defined as those <1.5 cm in diameter. They are seen in about 1 % of newborns. Small CMNs usually present as solitary, well-demarcated, light tan to dark brown uniformly colored macules or slightly raised papules. They can occur in any cutaneous location and be clinically indistinguishable from the so-called common acquired nevi [41].
According to published figures, small and medium-sized CMN carry a lifetime risk of malignant transformation of 2–5 %, while giant CMN carry a 4.5–10 % lifetime risk [42, 43]. However, there is ample variation of this estimated risk, and in general there is a tendency to exaggerate it. A systematic review of the literature published by Krengel et al. [44] showed an overall risk of melanoma of 0.7 %. Melanomas developing in small to medium-sized CMN generally begin to appear around puberty, and their incidence increases throughout adult life. In these nevi, MM develops at the dermoepidermal junction as melanoma in situ. Malignant transformation of giant CMN generally occurs in the deep dermal component of the lesion rather than in the dermoepidermal junction [25] (Fig. 13.4). Higher risk of malignant transformation correlates with larger lesional diameter, increasing number of satellite nevi, and location on the posterior axis [45].
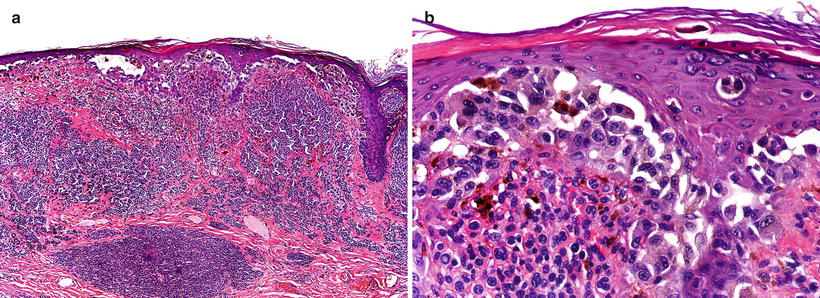
Fig. 13.4
(a) Malignant melanoma arising in the dermoepidermal juntion in a medium size CMN. (b) High power magnification of the juntional area
CMN may exhibit distinctive histologic features that help in distinguishing them from common acquired melanocytic nevi. Nevertheless, these features are not entirely pathognomonic for their diagnosis, but they are suspicious for congenital onset in the absence of documentation of the presence of the nevus at birth.
Histologic features, which are shared by small and giant congenital nevi (Fig. 13.3), include (1) the presence of nevomelanocytes within the lower two thirds of the dermis and within the subcutaneous tissue; (2) nevomelanocytes splaying or extending between the collagen bundles of the reticular dermis as single cells, or cords of cells; (3) extension of nevomelanocytes around and within hair follicles, sebaceous glands, eccrine apparatus, vessel walls, and nerves; (4) a perivascular and perifollicular distribution of nevomelanocytes simulating an inflammatory reaction such as figurate erythema; and (5) arrector pili that may be enlarged, distorted, and infiltrated by nevomelanocytes [41, 46–48].
The optimal management of congenital nevi is controversial. It is generally accepted that complete excision of the entire lesion in early childhood decreases the risk of malignancy, but this is difficult to accomplish in giant CMN, the lesion of highest risk. An alternative option is close monitoring by physical examination with serial follow-up and prompt excision if the lesion develops suspicious changes [34].
Additional complications associated with congenital melanocytic nevi, especially in patients with either giant or multiple nevi, include the spectrum of lesions described under the heading of neurocutaneous melanocytosis (NCM), which encompass proliferations of neural crest-derived melanocytic elements within the meninges and/or the brain, frequently accompanied by Dandy-Walker malformations, cysts of the IV ventricle, and other malformations of the central nervous system. Patients with this spectrum of disorders have variable outcomes depending on a number of factors, particularly the degree of CSF obstruction and the presence of associated CNS symptoms [37].
Clinical Presentation
MMs in prepubertal children are so rare that they are not usually suspected. Clinical findings of MM in children, particularly in adolescents, are similar to those seen in adults. Atypical morphological features are more frequently seen in children. Congenital MMs are also very rarely seen, can present de novo [49], in neonates with giant CMN [50], and may be secondary to transplacental metastasis from an affected mother [51].
In an early series of 125 patients with pediatric melanoma, the most common clinical presentations included increasing size of a mole, bleeding, color change, itching, palpable adenopathy, and palpable subcutaneous mass [52]. Compared with adult melanoma, a significant proportion of pediatric MM are amelanotic (50 %) or have a nodular configuration (30 %), and present at a greater median thickness (3.5 mm) [53].
The ABCDE clinical rule (Asymmetry, Border irregularity, Color variability, Diameter >6 mm, and Evolution) that is often used in identifying potential adult melanomas may be difficult to apply to pediatric skin lesions, because common pediatric lesions such as Spitz or benign nevi can have these features. Pediatric patients are also more likely to present with amelanotic lesions, not a common feature in adult melanoma [53] (Fig. 13.5).
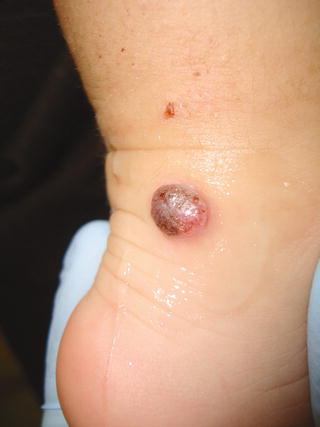
Fig. 13.5
Amelanotic melanoma presenting de novo in the leg of an infant (Courtesy Dr. A. Lassaletta, Madrid)
Most patients (>80 %) present with localized disease at diagnosis; the remainder have either regional lymph node disease (10–15 %) or distant metastasis (1–3 %). Any organ may be involved by metastasis, including lung, liver, lymph nodes, subcutaneous tissue, and brain [30].
Histopathology
The histopathological criteria used for adult patients should also be used for pediatric melanomas (Table 13.3). The most useful features distinguishing MM from nevi are large size (>7 mm), ulceration, high mitotic rate (>4 mitoses/mm2), mitoses in the lower third of the lesion, asymmetry, poorly demarcated lateral borders, lack of so-called maturation, finely divided melanin, and marked nuclear pleomorphism [54, 55].
Table 13.3
Histological features of malignant melanoma
Histopathology |
• Asymmetry • Ill-defined borders • Lack of maturation • Atypia of melanocytes (variable) • Mitotic figures may be present • Irregular pagetoid spread • Lentiginous junctional proliferation • Ulceration and/or “consumption of the epidermis” • Predominance of solitary units of melanocytes • Marked confluence of melanocytes along the dermal-epidermal junction • Marked dyscohesion of melanocytes • Lichenoid inflammatory reaction may be present • Fibrosis and dermal regression may be present |
Immunophenotype |
• Positive for S100, Melan-A, HMB-45, tyrosinase |
Variants |
• Common MM (superficial spreading, nodular, acral lentiginous) • Small cell melanoma • Spitzoid Melanoma |
Differential diagnosis |
• Spitz nevi • Atypical spitzoid neoplasm • Reed nevi • Proliferative nodule in CMN • Acquired or congenital nevi with pagetoid melanocytosis and lentiginous melanocytic proliferation |
Melanoma in children is classified in three groups [19], as per the following sections:
Conventional melanoma. Most melanomas in children can be included in this group. Of the four classical histologic subtypes (superficial spreading, nodular, acral-lentiginous, and lentigo maligna), superficial spreading melanoma is the most common type in both pediatric and adult patients [34] (Fig. 13.6). Young patients appear to have a greater frequency of nodular melanoma. Melanomas of glabrous skin are exceedingly rare in childhood. Lentigo malignant melanoma does not occur in childhood, suggesting that sun exposure plays a less important role in the development of melanoma in young people [56].
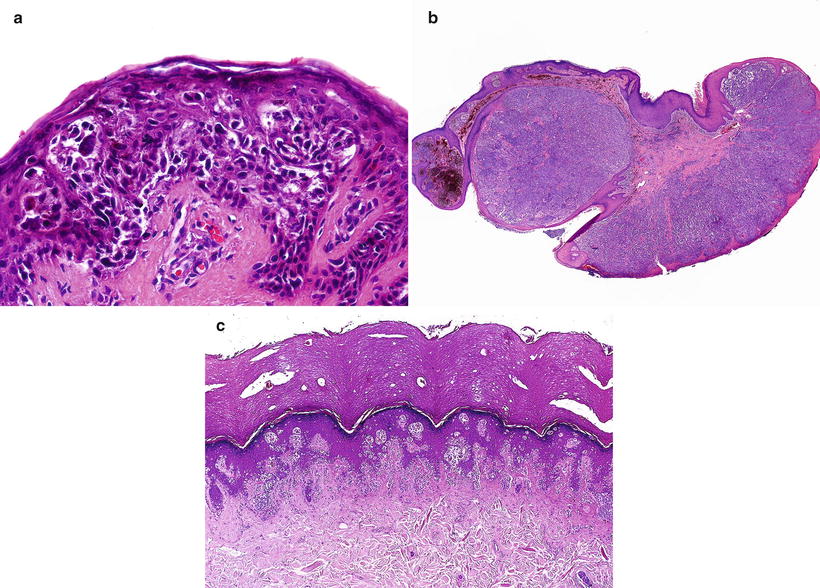
Fig. 13.6
Conventional or adult type malignant melanomas in childhood. (a) Superficial spreading melanoma. (b) Nodular melanoma. (c) Acral lentiginous melanoma (Courtesy Prof. L. Requena, Madrid)
Small-cell melanoma. These tumors may appear de novo or develop in a congenital nevus, are frequently localized in the scalp, show striking Breslow thickness, and are associated with a fatal outcome in most patients. Small-cell MMs are comprised of monomorphous cells arranged in sheets or in organoid configurations. The high cellular density, lack of maturation, and high mitotic rate are the clues to recognize this lesion [54, 55] (Fig. 13.7).
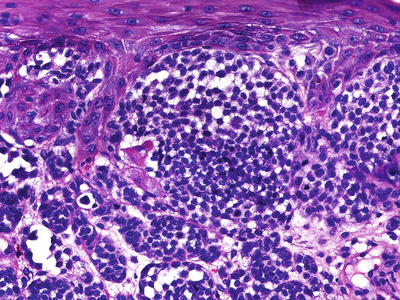
Fig. 13.7
Small-cell melanoma, containing monomorphous cells arranged in sheets
Spitzoid melanoma. Some melanomas may exhibit features suggesting a Spitz nevus, such as large epithelioid cells and spindle cells arranged in fascicles, epidermal hyperplasia, wedge-shaped configuration, and epidermal clefting surrounding intraepidermal nests. Although most of these lesions also show unequivocal features of a malignant melanocytic neoplasm (Fig. 13.8), some of them may represent extreme difficulty under the microscope, and require additional molecular analyses for definitive discrimination (vide infra).
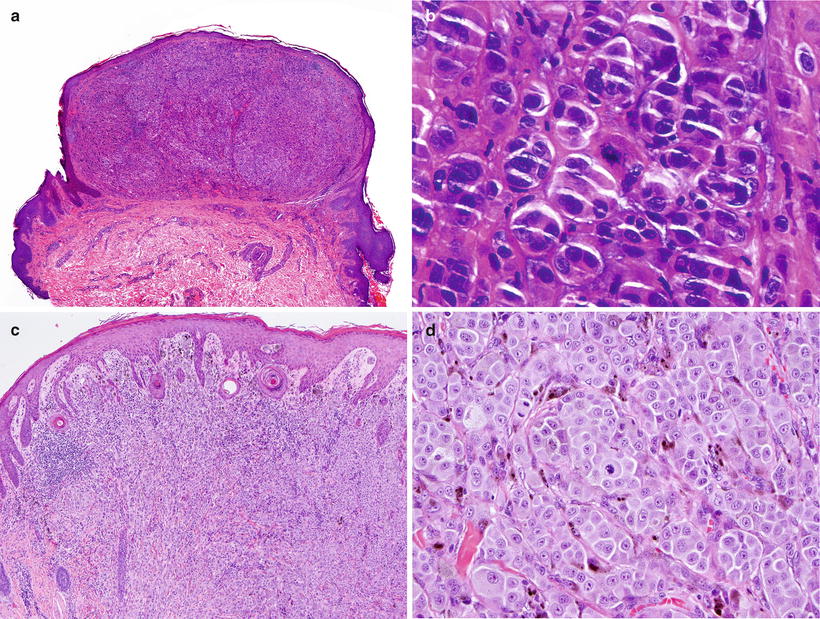
Fig. 13.8
Spitzoid melanoma. (a) A symmetric and well-delimited lesion that mimics a Spitz nevus at low power magnification. (b) The melanocytes have an epithelioid appearance, but there is no “maturation” and mitotic figures are present at the deepest part of the tumor. (c) Low power view of another example with epithelioid elements filling the dermal compartment. (d) The cells are highly atypical and feature increased mitotic activity (Courtesy Prof. L. Requena, Madrid)
Differential Diagnosis
Pagetoid melanocytosis and lentiginous melanocytic proliferations are commonly observed in acquired or congenital nevi developing in children, particularly in the glabrous skin. These changes must not be overinterpreted, unless architectural disorder and cytological atypia are prominent.
The main differential diagnoses of MM in children are Spitz nevus, atypical spitzoid neoplasms, nevus of Reed, and proliferative nodules in CMN.
Spitz Nevus
Spitz nevi (SN) are benign proliferation of large spindled, oval, or large round (epithelioid) melanocytes that begin in the epidermis and evolve into compound or intradermal stages [19].
SN usually present as a single, dome-shaped papule or nodule with a diameter of 6 mm or less (Fig. 13.9), but larger lesions are not uncommon. Most examples occur on the face and head in children or lower extremities in young adults, particularly women. Their color may vary from nonpigmented through pink to red-brownish and even black. SN commonly appear suddenly and grow rapidly for a period, after which they plateau and remain stable. However, color changes, bleeding, and pruritus may occur. Unusual variants include grouped or agminated [57–60], disseminated SN [61], and eruptive [62].
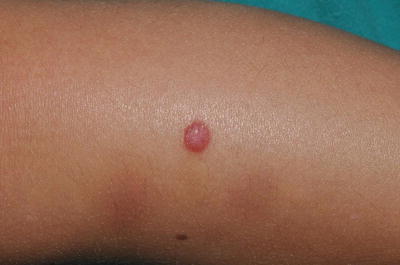
Fig. 13.9
Typical Spitz nevus presenting as a dome-shaped pink papule on the leg. These lesions may be clinically misdiagnosed as hemangiomas
The classic SN (Fig. 13.10) is dome shaped and symmetric with abrupt attenuation of the junctional nests at the lateral borders of the lesion. The nevus is composed of variable proportions of spindle and epithelioid nevomelanocytes, plump and with abundant cytoplasm, featuring a centrally located vesicular nucleus, often with prominent nucleolus. The proliferating cells sometimes show bizarre shapes, and multinucleated cells may be present. The cytoplasm of the epithelioid cells may have a ground glass appearance, and melanin is usually absent. If present, melanin is symmetrically distributed and absent at the deepest level of the lesion.
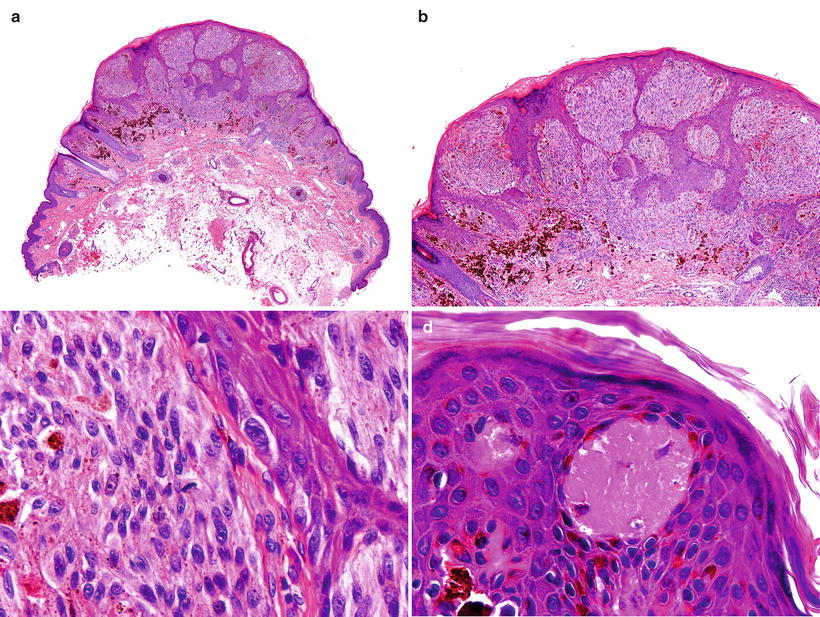
Fig. 13.10
Histological features of the Spitz nevus. (a) A well-circumscribed lesion (b) composed of spindle and epithelioid cells (c) with some superficial mitotic figures and (d) Kamino bodies
A common feature in SN is the presence of Kamino bodies, which are eosinophilic and globular deposits of basement membrane material found within the epidermis, usually above dermal papillae and present in variable numbers from case to case [63]. SN usually features a compound histological pattern, although junctional and intradermal lesions are not uncommon. Compound SN are symmetric, well circumscribed, and often wedge-shaped, with large nests of nevomelanocytes in the epidermis and in the underlying dermis. The proliferating cells are relatively uniform in size and shape, typically oriented perpendicular to the epidermis. Artifactual clefting around the nests is usually seen. Pagetoid spread of nevomelanocytes can be present, mainly in the lower half of the epidermis. This process is usually confined to the center of the lesion, but it can be quite marked in developing junctional SN in young children [64]. SN show “maturation,” traditionally defined as progressive reduction of melanocytic nests and cellular size from the top to the bottom. Nevomelanocytes in the dermis are mostly nested or arranged in fascicles and do not form sheets. There are often single cells infiltrating between collagen bundles in the reticular dermis at the base of the lesion. Non-atypical mitotic figures may be seen, but they are usually not numerous, occur in the mid- to upper portion of the lesion, and are more prevalent in the compound type of SN compared with the junctional and intradermal SN [65]. Some authors suggest a cutoff number of up to 2 mitoses per lesion for benign SN [66]. However, numerous mitoses can be present in rapidly growing SN and recurrent, regressing, or traumatized ones [67].
The epidermis associated with SN is acanthotic, hyperplastic, hypergranulotic, and hyperkeratotic. Perfectly symmetric epidermal hyperplasia with expansion at the center and attenuation at the periphery is a feature of SN, whereas there is usually “consumption of the epidermis” in spitzoid MM.
Lymphocytic inflammatory infiltrates are often seen around vessels and at the base of the lesion but not admixed with the nevomelanocytes. Blood vessels may be dilated and prominent. Variants of SN include desmoplasic, angiomatoid, myxoid, plexiform, and rosette-like types.
There are only a few histological differences between SN in children and in adults. Kapur et al. [68] compared 27 features in specimens obtained from both children and adults. The adult lesions were significantly more likely to be intradermal, and to display dermal fibroplasia, but otherwise to show histological similarity to their pediatric counterparts. Requena and colleagues found hyalinization to be the only histologic parameter of statistical significance more frequently encountered in adults than in children [65].
Although distinction between SN and spitzoid MM cannot be reliably made on the basis of immunohistochemistry, some stains may be useful in distinguishing spitzoid melanocytic neoplasms from nonmelanocytic tumors. HMB-45 and tyrosinase diminish toward the base of SN, in contrast with the diffuse pattern seen in spitzoid MM. MIB-1 staining is higher in MM than in SN (29.7 and 4.0 %, respectively). As S100 and Melan A are diffusely expressed in both SN and spitzoid MM, they are only useful in differentiating SN from nonmelanocytic tumors [69–73].
CGH and FISH are promising ancillary tests, and a large body of literature on this topic is rapidly accumulating [74]. The sensitivity and specificity of these techniques is improving fast although there are still some pitfalls, including false positive and false negative results, and the fact that some histologically “borderline” lesions show “borderline” cytogenetic features as well. Among the newer and most useful approaches is the use of a FISH probe set including 9p21, 6p25, 11q13, and 8q24, which has recently been demonstrated to reach a sensitivity of 94 % and a specificity of 98 % [75].
Atypical Spitzoid Neoplasms
This is a poorly defined and likely heterogeneous group with morphological overlap between melanoma and Spitz nevi. In contrast to classic SN, atypical spitzoid neoplasms (ASN) exhibit more worrisome features, such as size >1 cm in diameter, ulceration, increased cellularity or prominent confluence of nevomelanocytes in the dermis, extension into dermis or subcutaneous fat, dermal mitoses, cytological atypia, and lack of “maturation” at the base. Using some of the histological criteria noted above in conjunction with clinical characteristics, Spatz et al. [76] developed a grading system that stratified ASN into low-, intermediate-, and high-risk groups based on total accumulated atypical features (Table 13.4).
Table 13.4
Grading system for risk of metastasis in atypical spitzoid neoplasms
Parameter | Score |
|---|---|
Age (years) | |
0–10 | 0 |
11–17 | 1 |
Diameter (mm) | |
0–10 | 0 |
>10 | 1 |
Involvement of subcutaneous fat | |
Absent | 0 |
Present | 2 |
Ulceration | |
Absent | 0 |
Present | 2 |
Mitotic activity (mm2) | |
0–5 | 0 |
6–8 | 2 |
≥9 | 5 |
Although ASN have generally been reported to have a good prognosis, well-documented cases of metastasis and death exist [77–79]. Because the malignant potential of these lesions is uncertain if the thickness is >1 mm, many clinicians and patients make decisions to treat them as if they were melanomas with excision and, often, a sentinel lymph node biopsy [80]. Newer genetic alterations are being described that will contribute to an improved understanding of these neoplasm. Among them, there is recent description of an autosomal dominant tumor syndrome with high penetrance for melanocytic tumors and distinct clinical and phenotypical features associated with germ line mutations of the BAP1 gene [81]. Individuals affected by these ASN show BRAF mutations and loss of BAP1 expression, changes also seen in 28 % (from a total of 32 cases studied) of sporadic ASN [82].
Nevus of Reed
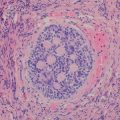
The nevus of Reed (NR) is a melanocytic proliferation histologically characterized by non-atypical, uniform, heavily pigmented spindled melanocytes [83, 84]. Although NR is a benign tumor, its natural history remains incompletely studied, and the lesion can be histologically or clinically misinterpreted as melanoma [84].
NR presents as a well-circumscribed, uniformly dark-brown or black lesion, averaging 3–5 mm in diameter (Fig. 13.11). Histologically, NR is symmetrical and shows a sharp lateral circumscription. The nevomelanocytes are arranged in vertical nests at the dermo-epidermal junction but can be seen above the dermal/epidermal junction, associated with confluence of the nests. The proliferation of nevomelanocytes may extend into the papillary dermis. Occasional mitoses may be found. Epithelioid nevomelanocytes are seen in a minority of cases. Commonly, the epidermis is slightly hyperplastic and shows marked hyperpigmentation of the basal keratinocytes. Kamino bodies can be observed in about half of the cases. An inflammatory infiltrate composed of lymphocytes and histiocytes with many melanophages is found within the papillary dermis [47, 84, 85] (Fig. 13.12).
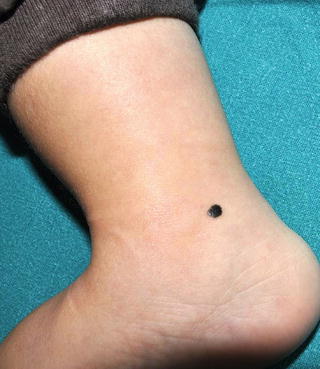
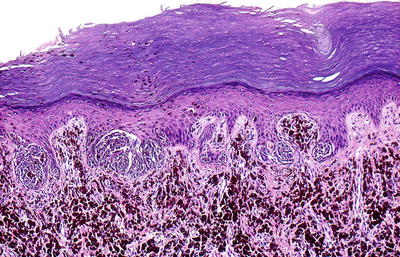
Fig. 13.11
Nevus of Reed. A sharply circumscribed black papule on the leg of a child (Courtesy Dr. A. Torrelo, Madrid)
Fig. 13.12
Histological features of the nevus of Reed. A junctional spindle cell neoplasm with vertically oriented nests and numerous dermal melanophages
There is significant controversy as to whether the NR represents a separate clinical entity or a variant of the classical Spitz nevus.
Proliferative Nodules in CMN
Proliferative nodules (PN) in congenital melanocytic nevi are defined as atypical melanocytic proliferations that predominantly manifest in the neonatal period within a pre-existing giant CMN [19]. The vast majority of atypical nodular proliferations developing in CMN are biologically benign and must be differentiated from MM arising in the deep dermal component of CMN.
PN present clinically as one or more dark brown to black plaques or nodules above a giant CMN (Fig. 13.13). The lesions may become lighter and show regression after years. In some cases, a palpable mass can be found deeply in the skin. Features that favor a benign lesion include blending of these cellular aggregates with the surrounding nevomelanocytes, low mitotic rate, absence of uniform high-grade cytologic atypia, and absence of inflammatory infiltrates or necrosis (Fig. 13.14a, b) [41].
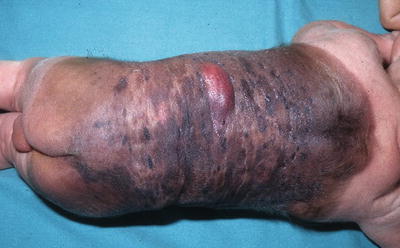
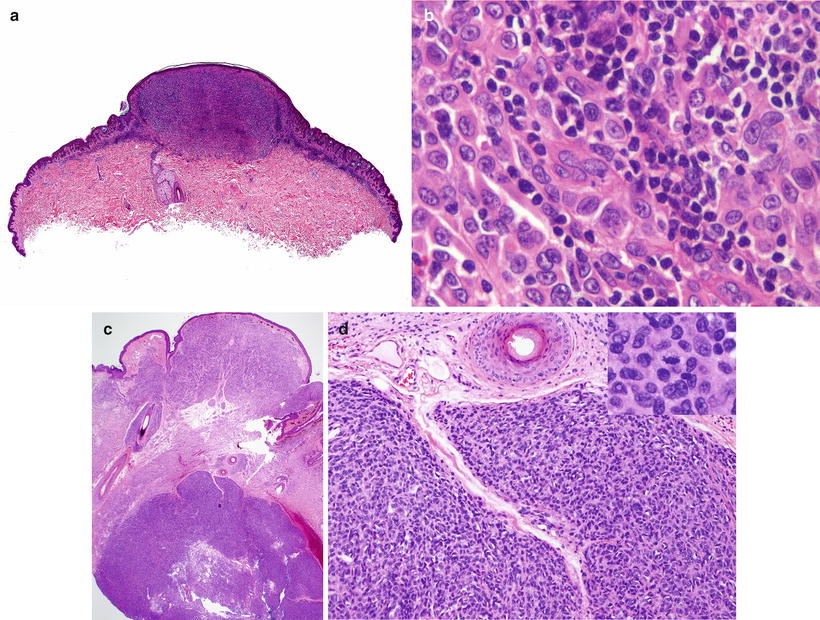
Fig. 13.13
Proliferative nodule arising within a giant congenital melanocytic nevus (Courtesy Dr. A. Torrelo, Madrid)
Fig. 13.14
Histological features of a proliferative nodule in a congenital melanocytic nevus. (a) Low power view. (b) The epithelioid non-atypical melanocytes merge with the smaller nevomelanocytes that can be recognized around the nodule. (c) An atypical proliferative nodule, sharply demarcated and with an expansile appearance, underlying an area of conventional giant congenital melanocytic nevus. (d) Cells in the atypical proliferative nodule show a compact arrangement and do not blend with the adjacent nevus cells. Mitoses are increased (inset)
Atypical PN appear sharply demarcated, show an expansile pattern of growth, and feature epidermal effacement, pleomorphism, and higher mitotic activity. They feature an increased Ki67 and PHH3 staining compared to non-atypical PN and the adjacent nevus cells surrounding the nodule. On this basis, some authors consider them as lesions intermediate between PN and MM. However, they show the same pattern of BRAF and NRAS mutations seen in PN and surrounding nevus cells [86] (Fig. 13.14c, d).
Prognosis and Treatment
Pediatric melanoma patients appear to have better prognosis than adults with melanoma even when matched for stage. Although this has not been thoroughly studied, it is conceivable that differences in clinical behavior reflect distinct tumor biology [27, 87].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


