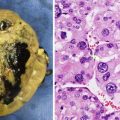
Fig. 15.1
MPSNT arising from ganglioneuroma. A ganglionic tumor with low grade schwannoma cell stroma segues into a high grade spindle cell neoplasm, X400
Patients with MPNST tend to be symptomatic, presenting with an enlarging mass, sometimes associated with neurologic symptoms (pain, sensory deficits, and weakness), dependent upon the underlying nerve involved. A rapidly enlarging mass that is mobile perpendicular to the course of a peripheral nerve or that yields a Tinel’s sign on percussion is highly suspect for MPNST [1]. Likewise any enlarging mass detected within a patient with NF1 should be considered MPNST until proven otherwise.
Imaging Features
Imaging Features of Malignant Peripheral Nerve Sheath Tumors (MPNSTs)
Before describing the imaging features of MPNST, benign PNSTs (neurofibroma and schwannoma) are briefly discussed. Distinguishing imaging features of malignant versus benign tumors are emphasized.
Neurofibroma
Neurofibroma is a non-encapsulated benign peripheral nerve sheath tumor that has been described in three forms: localized, diffuse, and plexiform [49, 50]. Approximately 90 % are of the localized variety; most are superficial affecting cutis and sub-cutis. The diffuse form is uncommon, primarily affecting children and young adults, involving the subcutaneous tissues of the head and neck region, trunk, and extremity, showing a plaque-like elevation of the skin and thickening of the sub-cutis and may extend to the fascia over muscle [51]. The majority of both localized and diffuse forms is not associated with neurofibromatosis type 1 (NF-1), also known as von Recklinghausen’s disease. However in the setting of NF-1, the neurofibromas tend to be larger, multiple, and deep in location. The plexiform form of neurofibroma is pathognomonic of NF-1, presenting usually in early childhood as a tortuous mass involving a long segment of a major nerve trunk and expanding into the nerve branches. It may be superficial or deep in location, exhibiting different MR imaging characteristics (see below). Fifty percent of plexiform neurofibromas occur in the head and neck, face, and larynx.
Plain Film Radiography
Plain radiography may show enlargement of neuroforamen when a dumb-bell shaped neurofibroma is involving a spinal nerve root.
Ultrasonography
High-resolution sonography of a neurofibroma shows a round homogeneous hypoechoic mass (Fig. 15.2) located centrally along the course of a peripheral nerve with distal acoustic enhancement, simulating a cystic lesion (pseudocystic appearance) [52, 53]. A sonographic target lesion with hypoechoic periphery and hyperchoic center may be seen, corresponding to myxomatous peripheral and fibrocollagenous central regions, respectively [53]. In diffuse neurofibromas, hyperechoic masses permeated by multiple interconnecting hypoechoic tubular or nodular structures have been reported in the subcutaneous fat zone. Differential diagnoses include cutaneous lymphoma, angiomatous lesions, cellulitis, and hemorrhage [54]. Sonography, even with duplex and color Doppler techniques, is not able to distinguish among neurofibromas, schwannomas, and malignant peripheral nerve sheath tumors [53, 55].
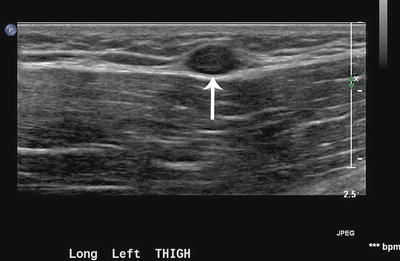
Fig. 15.2
Neurofibroma of left thigh in an 18-year-old boy. Longitudinal view of the thigh shows a focal hypoechoic mass (arrow) in the deep subcutaneous plane against the echogenic muscle fascia with homogeneous echotexture. Excision biopsy showed neurofibroma
Computed Tomography
On CT, localized neurofibromas appear as a well-defined hypodense (compared with muscles) mass from the presence of Schwann cells (fat content of myelin), myxoid tissue (high water content), entrapment of fat, and cystic areas of hemorrhage or necrosis [56]. After intravenous contrast injection, over half of neurofibromas show little or no contrast enhancement (Fig. 15.3). Visible bony erosions associated with dumbbell tumors are seen with CT [57].
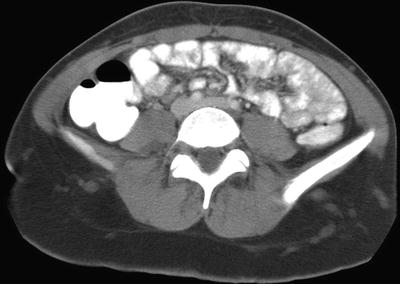
Fig. 15.3
Diffuse neurofibromas in a 14-year-old girl with neurofibromatosis type 1. Contrast-enhanced axial CT image of the pelvis shows a network of interconnecting soft tissues masses in both gluteal subcutaneous fat due to multiple neurofibromas
Magnetic Resonance Imaging
On MRI, a neurofibroma is spindle or ovoid in shape and is in contiguity with a specific nerve [58]. It shows low to intermediate signal intensity (or isointense to adjacent muscles) on T1W images and high signal intensity on T2W images. The high signal intensity on T2W images may be homogeneous or showing a characteristic “target sign” with a hyperintense periphery and a hypointense central region [59–61]. The high peripheral signal is related to myxoid and water contents and the central low signal is related to dense collagen and fibrillary tissues. It is important to use a wide window setting to allow demonstration of this sign [62]. After intravenous contrast, enhancement is inhomogeneous in two-thirds of cases and uniform in the rest. A neurofibroma is typically fusiform in shape with tapering ends contiguous with the parent nerve. When large, the tumor has a fascicular appearance (fascicular sign). A “split-fat” sign has been described when a neurofibroma originating from the nerve in an intramuscular location is surrounded by a rim of fat. Muscle atrophy may be seen in the muscle supplied by the nerve with neurofibroma. The diffuse form of neurofibroma presents as an ill-defined network of interconnecting neurofibromas extending through the involved subcutaneous tissue, showing low signal intensity on T1W images, high signal intensity on T2W images, and significant intravenous contrast enhancement on MR imaging. Prominent internal vascularity is common [51]. Plexiform neurofibromas, when deep, appear as hypodense multilobular masses within a major nerve distribution on CT scans and large conglomerate of masses of neurofibromas on MR imaging (Fig. 15.4). When superficial, plexiform neurofibromas in patients with NF-1 tend to be unilateral, have nontarget signal characteristics, exhibit a diffuse and infiltrative morphology, extend to the skin surface in a branching reticular fashion with small fascicles or nodules (Fig. 15.5), and can be mistaken for venous malformation by MR imaging [63–65].
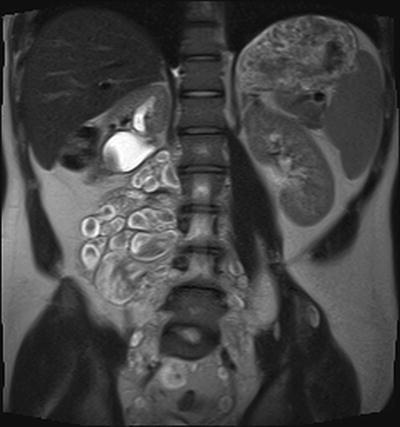
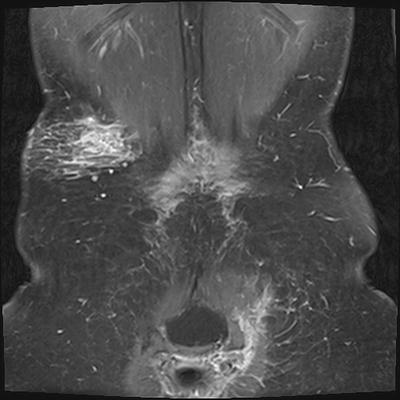
Fig. 15.4
Deep plexiform neurofibromas in a 17-year-old boy. Coronal HASTE MRI imaging of the abdomen and pelvis shows a large conglomerate of masses in the right psoas muscle, most showing “target sign”. Partial excisional biopsy of some of these masses shows “neurofibroma without evidence of malignant change”
Fig. 15.5
Superficial plexiform neurofibromas in a 19-year-old female with neurofibromatosis type 1. Post-contrast coronal fat-saturated T1W MRI shows a diffuse infiltrative and reticular branching pattern of plexiform neurofibromas in the right flank and left gluteal regions
Schwannoma (Neurilemmoma)
Schwannoma is an encapsulated (within the epineurium) nerve sheath tumor presenting as a slowly growing soft tissue mass involving nerve trunks in limbs, head and neck, posterior mediastinum, and retroperitoneum [49, 66]. The mass is usually painless unless large enough to compress the adjacent nerve. Those associated with NF-1 are usually multiple or plexiform. Schwannomas associated with neurofibromatosis type 2 (NF-2) tend to be central in location [57]. Larger lesions may undergo cystic degenerative changes, hemorrhage, calcification, and fibrosis.
Plain Film Radiography
Plain radiography usually does not reveal the mass itself, but may show scalloping of bone adjacent to the tumor.
Ultrasonography
Ultrasonography may show, especially in large nerves, a mass eccentrically positioned with respect to the affected nerve. However, there may be limitations, even with meticulous scanning technique, in demonstrating this eccentric positioning in up to 40 % [55].
Computed Tomography
On CT scan, schwannoma appears as a well-defined isodense or hypodense mass compared to muscles and shows enhancement after IV enhancement except in necrotic areas. Scalloping of the adjacent bone and expansion of the spinal canal are characteristic features on CT (Fig. 15.6a, b).
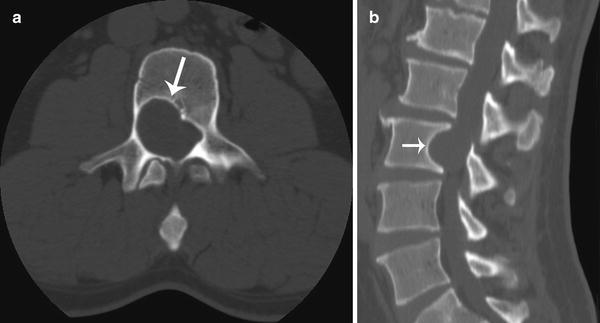
Fig. 15.6
Intraspinal schwannoma in a 33-year-old adult male with neurofibromatosis type 1. Axial (a) and sagittal (b) CT images of the lumbar spine show scalloping of the posterior right vertebral margin (arrows) at L3 level and expansion of the spinal canal
Magnetic Resonance Imaging
MR imaging findings are similar to those described in neurofibromas, including a mass with a fusiform, spindle, or oval shape, in contiguity with a specific nerve, and showing a target sign, and “split-fat” sign. A mass eccentrically positioned in relation to the nerve and showing heterogeneity with cystic degenerative changes suggests a schwannoma [67]. A low signal peripheral rim (epineurium) is seen in 70 % of schwannomas versus in 30 % of neurofibromas [68–70]. The target sign in schwannoma is attributed to a central area of more cellular Antoni type A neurilemmoma and to a peripheral rim of hypocellular Antoni type B neurilemmoma [71].
Malignant Peripheral Nerve Sheath Tumor (MPNST)
Malignant peripheral nerve sheath tumor is a spindle cell sarcoma arising from a peripheral nerve or its attendant sheath or from a benign PNST [50]. It does not include tumor arising from the epineurium or the vasculature of the peripheral nerves [72]. Typically MPNSTs arise from preexisting plexiform neurofibromas. However it has been documented that 36 % of 34 MPNSTs from a cohort of 1475 NF-1 patients developed MPNSTs without a history of plexiform neurofibromas [73]. While 30-50 % of MPNST are associated with NF-1, only approximately 2–5 % of NF-1 patients develop MPNST [49, 67, 74–76]. Most tumors arise from nodular plexiform tumors associated within major nerve trunks (such as brachial plexus sacral plexus, and sciatic nerve) and patients tend to be symptomatic presenting with pain, sensory deficits, and weakness. MPNSTs metastasize to the lung, liver, brain, regional lymph nodes, bone and soft tissues, skin, and retroperitoneum, and carry a poor prognosis [76].
Plain Film Radiography
Although often normal, plain radiography may show a soft tissue mass (Fig. 15.7a), secondary changes in adjacent bones (erosion or overgrowth), and is essential for evaluation of metastases to chest (lungs and pleura). Calcification (chondroid, osteoid, or amorphous) is uncommonly seen.
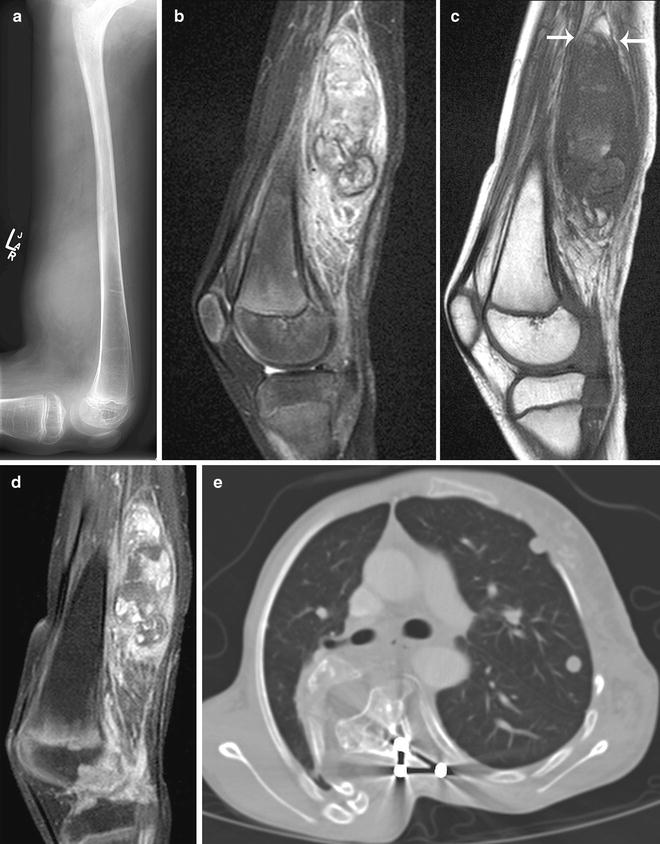
Fig. 15.7
Neurofibromatosis type 1 with neurofibroma progression to metastatic malignant peripheral nerve sheath tumor (MPNST). (a) Lateral plain radiograph of the thigh shows a soft tissue mass at distal posterior thigh. MRI of the thigh shows a large spindle-shaped posterior thigh mass with (b) inhomogeneous high signal intensities on fat-saturated T2W image, (c) inhomogeneous low signal intensities and “split-fat” sign (arrows) on T1W image, and (d) inhomogeneous enhancement on post-contrast fat-saturated T1W image. Biopsy of the mass showed a poorly differentiated MPNST arising within a neurofibroma. (e) Axial CT image shows multiple lung metastases
Ultrasonography
Sonography shows inhomogeneous hypoechoic masses that may have areas of hemorrhage, necrosis and calcifications [50]. A sonographic target sign is not present [53]. Duplex Doppler sonography shows a hypervascular pattern with corkscrew neovasculature, high velocities, and variable spectral waveforms [77].
Computed Tomography
Multi-detector CT (MDCT) may show abdominal primary tumor and metastases to the abdomen. On CT, MPNSTs are hypodense and ill-defined in outline with marginal enhancement after IV contrast medium.
Angiography
Angiography may show increased vascularity with characteristic corkscrew vessels at both ends of the tumor due to hypertrophy of the nutrient blood supplies to the nerve [71].
Magnetic Resonance Imaging
MPNSTs share MR imaging findings described above with benign peripheral neurogenic tumors. The involvement of the entering and exiting nerves results in the spindle shape of most MPNSTs . Although MRI can determine the site, extent, and change in size of plexiform neurofibromas, it does not reliably determine malignant changes [76, 78–80]. While any neurofibroma that rapidly increases in size in patients with NF-1 should be viewed with suspicion for malignant transformation, the growth rate of plexiform neurofibromas that give rise to MPNSTs is not a reliable predictor of malignancy. There may be periods of rapid growth, especially in adolescence, followed by periods of relative inactivity [81, 82]. Features that are concerning for malignancy include: tumor size over 5 cm in diameter, ill-defined margin, heterogeneity, intratumoral cystic changes, fat plane invasion or infiltration, absence of target or fascicular sign [59, 61, 62, 83], peritumoral edema, presence of peripheral enhancement, and history of having MPNST or previous radiation therapy (Figs. 15.7b–d, 15.8a–d). Only evidence of metastases (such as lung, pleura, bone, retroperitoneal node, and bone) is definitive for diagnosis of MPNSTs (Fig. 15.7e). While the split fat sign was present in 76.5 % of benign and larger PNSTs, only 33.3 % of MPNSTs showed this sign [58]. Whole body (head to feet) MRI with several table movement steps has been used in assessing the benign tumor burden in patients in NF-1 [84].
Nuclear Medicine Imaging
Bone scintigraphy may show findings indicating increased vascularity or mineralization and identify sites of bony metastases.
FDG-PET has recently proven to be useful in detecting metastatic and recurrent disease [85] and in the differentiation between benign and malignant peripheral nerve tumors using qualitative and semiquantitative SUVmax (maximum standardized uptake value) assessment [86, 87] (Fig. 15.8e–g). In patients with NF-1, FDG-PET is 95 % sensitive in the detection of MPNSTs. Because of the overlap in SUVmax for benign (ranging from 0 to 5.3, mean 1.5 ± 0.37) and malignant (ranging from 3.8 to 13.0, mean 8.5 ± 0.63) lesions, the addition of PET using 11C-methionine (measuring amino acid transport rate, protein synthesis, and cell proliferation in malignant tissues) has been found to improve specificity from 72 to 91 %[86]. In another series [87], no MPNSTs were detected with an SUVmax <2.5 and a small number of benign tumors had an SUVmax >3.5. Both benign and malignant peripheral nerve tumors had SUVmax between 2.5 and 3.5. The authors recommend that symptomatic neurofibromas with SUVmax ≥3.5 should be excised, and lesions with SUVmax between 2.5 and 3.5 should be reviewed clinically. In another study, Son and colleagues found varying degrees of FDG uptake in a patient with multiple benign neurofibromas on PET-CT and concluded that a low SUVmax may indicate benignity, but a high SUVmax does not always indicate malignancy [88]. Using ROC analysis, Warbey et al. found a significant difference in SUVmax between early (90 min) and delayed (4 h) imaging and between tumor grades, and recommended using a cutoff SUVmax value of 3.5 on delayed imaging to achieve maximal sensitivity in diagnosing MPNST [89]. The overlap of SUVmax between different tumor grades, however, did not allow accurate prediction of grade on an individual basis. The authors suggest that tumors with an SUVmax in the 3.0–3.5 range should be clinically reviewed at multidisciplinary and multi-specialist management meetings. In another ROC analysis [90], using SUVmax thresholds of 4.5, 6.1, and 8.5 were associated with sensitivities of 100, 94, and 65 % and specificities of 83, 91, and 100 %, respectively, for detecting malignancy. The authors also found that benign schwannomas are less reliably distinguished from the MPNSTs based on the SUVmax. In a pediatric series of NF-1 and plexiform neurofibromas, Tsai found that the SUVmax of typical and atypical plexiform neurofibromas (2.49 [SD = 1.50]) was significantly different from MPNSTs (7.63 [SD = 2.96]). Using an SUVmax cutoff value of 4.0, sensitivity and specificity were 100 and 94 %, respectively for distinguishing plexiform neurofibromas and MPNSTs [91]. Nodular target lesions seen on MRI in patient with NF-1 and plexiform neurofibromas were found to have increased FDG uptake similar to that of MPNSTs, although they might be benign on biopsy [92]. Careful longitudinal clinical and imaging monitoring were recommended using MRI and FDG-PET to identify lesions of greatest concern to be biopsied. FDG-PET imaging can help in guiding targeted needle core biopsy of PNSTs [90], directing biopsy to the more metabolically active areas of the tumor. A newer tracer with 18 F-thymidine, which detects DNA turnover, may be useful in distinguishing low grade MPNSTs from active benign plexiform neurofibromas [76].
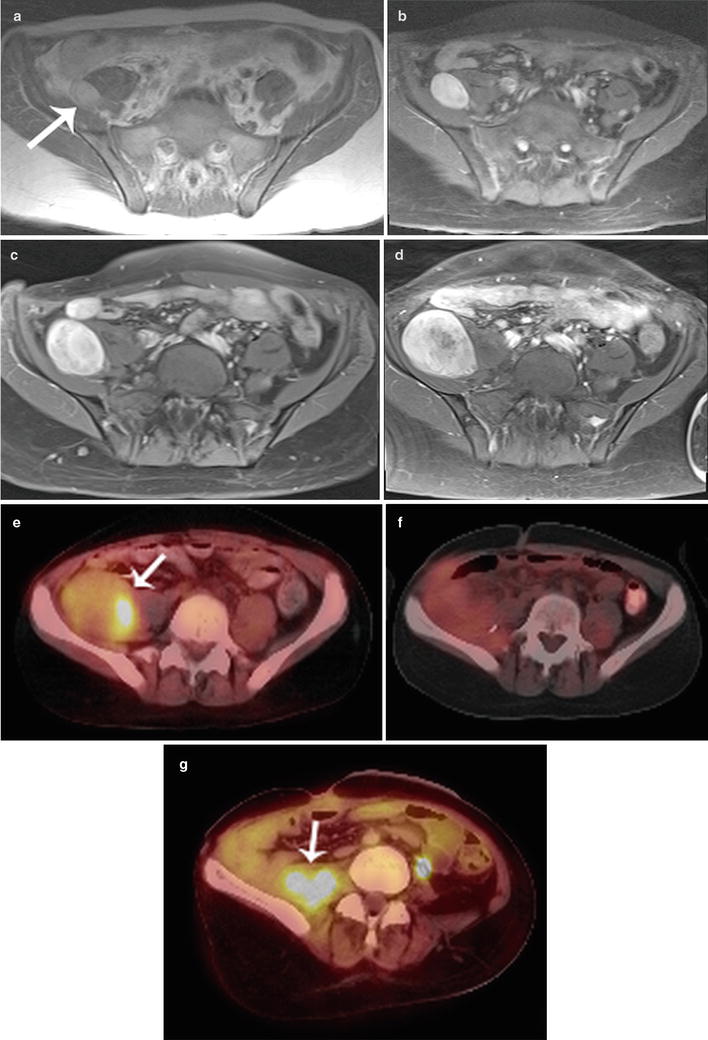
Fig. 15.8
Progression of neurofibromatosis type 1 first diagnosed in a 14-year-old girl over 5 years into a MPNST. Note progressive increase in size and inhomogeneity of enhancement of a right iliac fossa mass (arrow) from (a) 2005 (b) 2007 (c), 2009 and (d) 2010. 18FDG-PET-CT scan in 2010 (e) shows a 4.5 × 6.7 cm mass with SUV max of 5.5 (arrow). Excisional biopsy revealed MPNST arising in neurofibroma. (f) Follow up 18FDG-PET-CT scan in 2011 shows no residual tumor (SUV max, 2.0). (g) Subsequent follow up in 2012 showed recurrent biopsy-proved MPNST with SUVmax of 5.6 (arrow)
Pathology
Gross and Microscopic Features
MPNST
The macroscopic appearance of MPNSTs is highly variable, ranging from large and ominous to subtle. Most will present similar to other soft tissue sarcomas as bulky fusiform or expansive tumors with variable infiltration into surrounding structures. They tend to be large tumors, averaging 6–10 cm in dimension, the majority >5 cm [2, 6]. Identifiable origination from a nerve may or may not be present. MPNSTs have a firm tan to grey interior with areas of hemorrhage and necrosis, similar to other aggressive sarcomas. A pseudocapsule typically represents tumor-infiltrated soft tissue, often with reactive features. At the other end of the spectrum, foci of MPNST arising within an underlying plexiform neurofibroma (malignant degeneration) may not be grossly visible at all, identifiable only at the microscopic level.
Classic / conventional MPNST may display a wide variety of architectural arrangements, frequently posing a significant diagnostic challenge given its resemblance to a number of other soft tissue tumors. The most frequent appearance is one of a hypercellular sarcoma with interwoven fascicles of spindle cells (Fig. 15.9) [93]. Other patterns include a fibrosarcoma-like herringbone pattern, hemangiopericytoma-like with staghorn vasculature, and alternating loose and dense cellular regions (similar to Antoni A and Antoni B regions in schwannomas) (Fig. 15.10) [2]. Perivascular condensation of tumor cells is another helpful feature [2]. Growth within nerve fascicles is also common, though invasion into surrounding tissues is typical. Individual tumor cells are spindle shaped with variable amounts of surrounding pink cytoplasm; nuclei tend to be wavy or indented with tapered ends [2]. Large zones of geographic necrosis are seen in over one half (Fig. 15.11), and mitotic figures are generally easy to find (Fig. 15.12), often numbering several per single high power field.
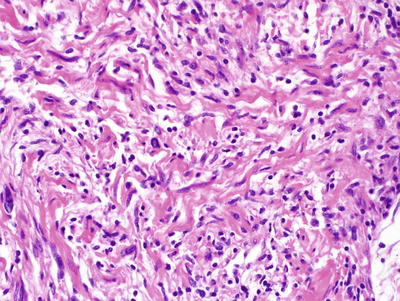
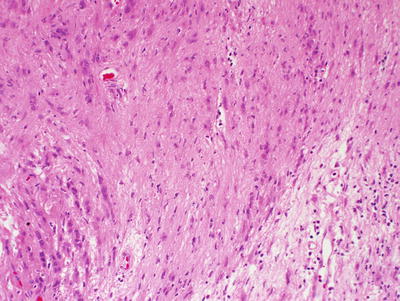
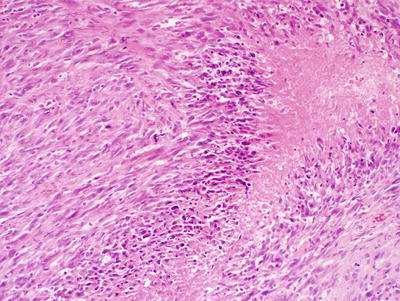
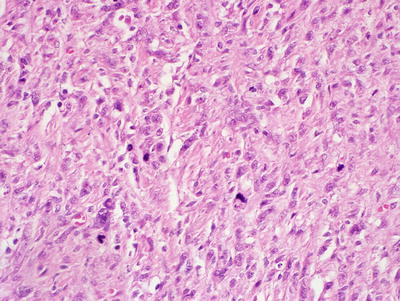
Fig. 15.9
MPNST, containing spindle cells arranged in interlacing bundles and containing tumor cells with hyperchromatic, enlarged nuclei and wavy contours, X400
Fig. 15.10
MPNST with contrasting hypercellular and hypocellular zones, analogous to schwannoma, X200
Fig. 15.11
MPNST with areas of geographic necrosis, X200
Fig. 15.12
MPNST with pronounced mitotic activity, X400
Fine needle aspiration cytology is being increasing employed in the diagnostic workup of soft tissue lesions, and our knowledge of the salient cytologic features of MPNST has become increasing refined over the past decade. Cytology aspirate smears of MPNST are typically hypercellular with a combination of cohesive cell clusters of variable size and cellularity together with numerous single tumor cells and naked nuclei [94, 95]. A fascicular pattern may be encountered in some cell clusters, though is not universally present; storiform or whorled patterns may also be encountered [30]. A fibrillary background may be present [95]. Individual tumor cells are spindle shaped with variable contour; they may be elongated with tapered ends, kinked, angulated, or comma-shaped [30, 94, 95]. Wavy nuclei, representing a quite helpful diagnostic feature when detected, are inconsistently present in MPNST cytology samples [94, 95]. Nuclear pleomorphism is a frequent finding, as are mitotic figures (including atypical forms) [30, 95]. A “dirty” necrotic background material may be seen [30].
The majority (85 %) of MPNSTs are high grade sarcomas. As such, they show evidence of hypercellularity, invasion of surrounding tissues (often with vascular invasion), together with nuclear pleomorphism, elevated mitoses (generally >5 mitoses per 10 high power field), ± necrosis [1]. It is the presence of necrosis that distinguishes high grade from intermediate grade tumors. Low grade MPNSTs make up the minority, appearing histologically similar to cellular neurofibroma, though having comparably increased cellularity and significant nuclear atypia (hyperchromasia and larger nuclear size) (Fig. 15.13a, b).
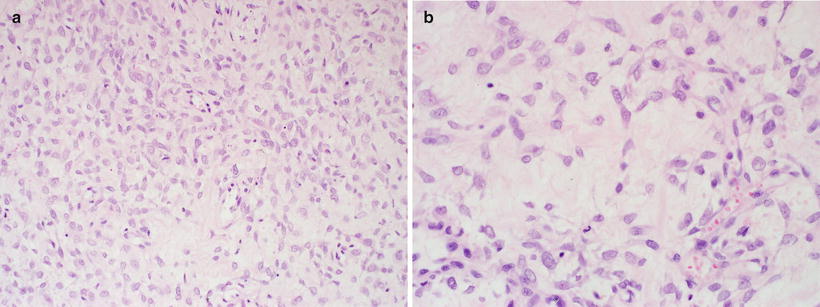
Fig. 15.13
Low grade MPNST containing hypercellular foci (a) amid zones of paucicellular neurofibromatous foci (b), X400. The latter exhibits cells with increased size and mild pleomorphism
Similar to low grade peripheral nerve sheath tumors, MPNSTs may show S100 positivity by immunohistochemistry; unfortunately S100 is detected in only 50 % of cases, and more often only scattered individual cells are S100 positive [18, 96–99]. Low grade MPNSTs tend to have more diffuse S100 positivity compared to their higher grade counterparts [98]. SOX10, a pan-schwannian marker, will be positive in up to 50 % of MPNST, but unfortunately nearly half of MPNSTs will be negative for both SOX10 and S100 [97, 100]. Interestingly, it has been demonstrated that MPNSTs have a heterogeneous cell composition, containing EMA and Glut1-positive perineurial cells, as well as CD34 positive endoneurial fibroblasts [99]. In some cases, cellular structures resembling tactoid bodies can be found (Fig. 15.14). Collagen IV is often present between individual tumor cells or cell groups, positivity tends to be focal and discontinuous [18]. Nestin, GFAP, Leu7, and NSE are demonstrable in some cases [96, 101]. A variety of cell cycle regulatory proteins may also be detected by immunohistochemistry, with low and high grade tumors showing different profiles. Whereas p16 and p27 tend to be positive in low grade MPNSTs, p16 and p27 expression tends to be lost in high grade MPNST, which instead often shows nuclear p53 expression [98, 102, 103].
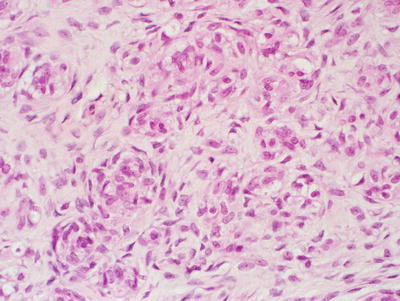
Fig. 15.14
MPNST with rounded, cellular structures resembling tactoid bodies, X400
MPNST Variants
Approximately 15 % of MPNSTs exhibit some form of divergent differentiation, harboring various mesenchymal or epithelial / epithelioid components [104]. Features of the most well-recognized of these variants are summarized below. It should be noted however, that a wide variety of heterologous elements may much more rarely be encountered; these include areas of fibroblastic [105] smooth muscle differentiation [93], or primitive neuroectodermal tumor (PNET)-like differentiation [106]. Examples of MPNST with pluridirectional differentiation have been described, bearing a mixture of two or more of the following malignant tissue types: epithelioid, rhabdomyoblastic, osteogenic, chondroblastic, lipogenic, and pigmented neuroectoderm [2, 18, 107–111].
Malignant Triton Tumor (MTT)
MPNST containing a rhabdomyosarcomatous component is termed malignant triton tumor (MTT); this represents by far the most frequent form of divergent mesenchymal differentiation within MPNST [16]. Some authors have found that MTT tends to occur in an older population then conventional MPNST (mean age in 5th decade for the former) [112], though others have not found this to be the case [113]. The majority of MTT arise within the context of NF1, they tend to be larger than conventional MPNST, and are more frequent in the head and neck region [112, 113]. Microscopically, the rhabdomyosarcomatous component is typified by rhabdomyoblasts with rounded eosinophilic cytoplasmic bellies, though more elongated cells with discernible cross-striations may be seen (Fig. 15.15) [114]. Immunohistochemical stains for desmin, myoD1, muscle specific actin (MSA), and myogenin are positive in this component [114]. Pluridirectional differentiation is present in a proportion of cases. Similar to conventional MPNST, the vast majority of MTT are high grade aggressive neoplasms.
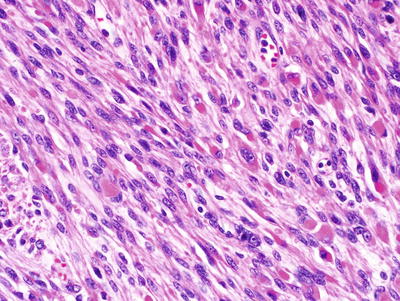
Fig. 15.15
Malignant triton tumor. The lesion consists of both neural and myogenous elements, the latter exhibiting abundant, eosinophilic cytoplasm and rounded contours, X400
Epithelioid MPNST
As the name implies, epithelioid MPNSTs contain variable proportions of cells with an epithelioid appearance. They tend to arise in either superficial or deep soft tissues of the extremities, often involving major nerves [2, 115, 116], though alternate sites have been documented as case reports [42, 117]. There is no association with NF1. In one instance, epithelioid MPNST was documented in the context of a precursor schwannoma, arising in a patient with schwannomatosis and germline SMARCB1 (Ini1) mutation [118]. Microscopically, these lesions have a nodular architecture, composed of cords and rows of rounded epithelioid cells with prominent nucleoli and brisk mitotic activity. They are diffusely positive for S100 and NSE by immunohistochemistry, while they lack more specific melanoma markers. Cytokeratin may be positive in some cases. Similar to other MPNSTs, collagen IV is frequently demonstrable between individual tumor cells and cell groups, corresponding to basement membrane material by electron microscopy [115, 116].
Epithelioid MPNST is not to be confused with glandular MPNST, a rare variant which in contradistinction contains well-formed glandular elements resembling benign intestinal-type epithelium. Similar to MTT, three quarters of these patients have underlying NF1, and pluridirectional differentiation is found in a significant proportion of these tumors [119]. In additional to markers typically positive in conventional MPNST, the glandular component is immunopositive for cytokeratin and CEA; chromogranin-positive neuroendocrine differentiation is often present [93, 119].
Perineurial MPNST (Malignant Perineurioma)
Less than 5 % of all MPNST will show evidence of perineurial differentiation, represented microscopically as a sarcoma containing spindle cells with elongated processes and a whorled to storiform architecture akin to that of benign perineuriomas. Similar to epithelioid MPNST, these tumors are not associated with NF1, nor do they appear to arise from neurofibromas [120]. They tend to present as large tumors involving the soft tissues of the extremities or trunk; nerve involvement is infrequent. In contrast to conventional MPNST, malignant perineurioma is negative for S100 but positive for EMA, vimentin, glut-1, and claudin-1 [99, 120, 121]. A small proportion will be positive for CD57 and/or CD34. Prognosis is comparably more favorable than that of conventional MPNST, though recurrence and distant metastases are not uncommon [120].
Molecular Diagnostic Features
Karyotypic analyses [122–124], and more recently array comparative genomic hybridization (CGH) [103, 125] of MPNSTs have indicated that the vast majority of these tumors harbor structural and numerical chromosomal aberrations. Balanced translocations are rare [124], whereas microsatellite instability may be detected in up to one third of cases [122]. The majority of MPNSTs (including both sporadic and NF1-associated) have gross inactivating alterations of the p16 (INK4A) gene on 9p21 [126–128]; though inactivation is mainly via deletions and rearrangements [128], promoter methylation may also play a role [127]. EGFR [129, 130], topoisomerase-IIα [131], neuregulin-1/erbB and insulin-like growth factor 1 receptor pathways [132, 133] have all been implicated in MPNST tumorigenesis, as have microRNAs mirR-204 and miR-21 [134, 135]. Gene expression profiling studies have indicated distinct molecular classes of MPNST, and have found overexpression of neural stem cell markers sox9 and TWIST1, and neuroglial differentiation-associated transcripts; these finding may be important for future targeted therapies [136–138].
Differential Diagnosis
Unfortunately, there are no specific histologic characteristics that allow for reliable differentiation of MPNST from other malignant sarcomas. Demonstration of origination from a peripheral nerve or underlying neurofibroma is clearly helpful, though is not always present. The fallback is therefore immunohistochemistry, which is used to help provide evidence of nerve sheath differentiation in MPNST and at the same time rule out other tumor types. Ultrastructural examination may be helpful in some cases, in demonstrating intratumoral basement membrane material, or the sarcomeric structures of MTT [114].
Differentiation of MPNST from monophasic synovial sarcoma (SS) can be particularly problematic as the later may involve nerves on occasion, and can express S100; MPNST can likewise rarely express EMA and low molecular weight cytokeratin [40, 93, 114]. Findings of cellular pleomorphism and CD34 positive cells would favor MPNST [40, 93] over SS. Sox10, if positive, may be useful as it represents a more reliable marker of neural crest origin then S100, and is typically negative in non-neural sarcoma which might otherwise be confused with MPNST [97]. TLE1 is a transcriptional corepressor overexpressed in synovial sarcoma; diffuse nuclear staining with anti-TLE1 antibody is seen in SS, whereas MPNST is typically negative [139]. Determination of SYT status by immunohistochemistry may be similarly helpful [140], being consistently positive in SS, paralleling the expected SYT-associated translocations demonstrable by FISH [141] or other molecular modalities; SYT alterations, in contradistinction, are not expected in MPNSTs. Leiomyosarcoma and solitary fibrous tumor are two other mimics; happily, immunopositivity for smooth muscle actin (SMA) and diffuse CD34 positivity, respectively, reliably separate these spindle cell neoplasms from MPNST.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


