(1)
Clinical Department, Urals Research Centre for Radiation Medicine, Chelyabinsk, Russia
Abstract
Chronic radiation syndrome is considered to be a system response of a human body as a unified whole to chronic radiation exposure. Human body is not a simple set of organs, and its response to total body exposure is not a sum of responses manifested by individual organs. Organs are functionally interconnected due to, first of all, the regulatory systems (nervous, endocrine, and immuno-hematopoietic), and this maintains the homeostasis. At the initial stage, CRS is a typical “disadaptive” pathology characterized by non-specific functional disorders of hematopoiesis, nervous system and internal organs. At the basis of such changes there are radiation-induced impairments in the regulatory systems. Visceral disturbances are secondary in nature, and they are reversible. If the exposure dose for certain organs exceeds the threshold values, then, not only functional, but also organic changes develop (vascular impairments, dystrophy, fibrosis, RBM hypoplasia, etc.), and the CRS course acquires an irreversible character.
It is well known that the experimental animals and people are able to tolerate higher doses of chronic than acute single exposure (Fliedner et al. 2002). This phenomenon is called radioadaptation. It is assumed that the mechanism of radioadaptation is associated with a set of compensatory-reparative response at the cellular, organ, and organismic levels. Tissue response to low-dose chronic exposure reflects the simultaneous development of cell damage and adaptive processes (Wolff 1996; Rigaud and Moustacchi 1996; ICRP 2012). Adaptive response of the cells are characteristic of low doses acute and chronic low dose rate exposures induced by low-LET radiation; they decrease with increasing dose, and at doses above 0.5 Gy, they are usually not observed (Fliedner et al. 2002). The data on the adaptation effects to high-LET radiation exposure are limited.
Increase in radioresistance is one of the radioadaptation manifestations, which is why the dose, at which no damaging effects occur, is significantly higher in case of the chronic rather than acute exposure (Smirnova and Yonezawa 2004). Radioadaptation preserves fertility, normal functional activity, and vitality of the individual during chronic exposure.
Radiation-induced cell and tissue reactions appearing after the exposure to high doses of IR are described best of all. The responses of complex biological systems to low doses differ in both nature and degree of manifestation from the expected responses per unit dose under high-dose exposure. As it became known in recent years, low doses of low-LET IR can induce a number of biochemical and functional cell and tissue responses that are not registered at high doses. It should be noted that understanding of CRS pathogenesis is of not only theoretical interest but also practical, as peculiarities of CRS pathogenesis determine the strategy of complex pathogenetic therapy, the main principles of which are not yet formulated. Unfortunately, up to the present moment the pathogenesis of many tissue reactions as well as chronic radiation syndrome in general is insufficiently studied.
3.1 The Most Essential Characteristics of Molecular and Cellular Mechanisms Involved in Radiation Effects Development
Lately significant progress was made in understanding the molecular genetic mechanisms of biological radiation effects. Results of the study of molecular and cellular response to radiation exposure, varying in nature, type, and dose of radiation, were published in UNSCEAR and ICRP reports (UNSCEAR 2009; ICRP 2012) and are undoubtedly of interest for understanding the pathogenesis, clinical manifestations, course, and outcomes of CRS. Due to the importance of these results but impossibility to discuss them in detail, it was necessary to formulate the main conclusions that are relevant to the issues raised in this monograph.
It is well known that ionizing radiation (IR) has an impact on biological systems at the molecular level, interacting with atoms and inducing energy deposition along the particle tracks. These events are more or less stochastically distributed throughout the exposed tissue and can affect genetic, structural, functional, and signaling components of the cell. They can damage molecules directly or indirectly, for example, through reactive oxygen species (ROS) emerging during the radiogenic hydrolysis. Damage of the signal pathways can be of particular importance, as they are involved in the amplification cascades, and control intercellular communication, integrating the functions of cells, tissues, and organs and of the whole body.
The probability of effects at higher levels depends on the type, quality, and extent of primary disorders of homeostasis at the molecular level and on radioresistance of different mechanisms controlling homeostasis. The latter exert antioxidant effect and are involved in the recovery of damaged molecules, particularly DNA, and in the removal of the cells with unrepaired damage from the tissues through apoptosis, cell differentiation, or by means of immune response.
The immediate response to the IR exposure and DNA damage is very well investigated. DNA double-strand breaks can be detected within a few minutes after exposure by means of immunohistochemical techniques. If the primary structural DNA damage was not lethal, they are usually repaired within 24 h. The process of DNA reparation depends on the individual characteristics. In mammals more than 150 genes are involved in this process.
In addition to the direct physiological barrier, an organism exhibits various adaptive reactions, especially in cases when the impairment of homeostasis is below the violation of the barrier, i.e., in a stressful situation. Adaptive reactions to oxidative stress are well known. Similarly, low dose rate exposure changes cellular signals through a temporary modification of the enzymes and hormones, which are involved in defense against ROS and reactive nitrogen species (RNS), in DNA synthesis, in DNA reparation, and in removal of lesions through different ways. Under the effect of IR, delayed up- or downregulation of physiological barriers can be observed at relatively low doses; adaptive reactions reach their peak at a dose of about 100 mGy. Adaptive reactions virtually disappear at doses exceeding 200 mGy and are not at all detected at 500 mGy (Joiner et al. 1996). The exception is apoptosis, the probability of which increases linearly with doses >500 mGy.
Radiation-induced changes in DNA indirectly influence complex physiological processes in an organism through transcriptional and other molecular, cellular, tissue, and body systems.
3.1.1 Physicochemical Process of the Interaction Between IR and Biological Tissue
The understanding of energy transfer along the tracks of IR, when they pass through the DNA molecules, has improved significantly in recent years, due to computer modeling using Monte Carlo method. Detailed information of IR interaction with different substances is provided in ICRP Publications 92 (ICRP 2003) and 99 (ICRP 2005). It is known that IR, while passing through the matter, gradually releases its energy, interacting primarily with electrons. The probability of its interaction with the nuclei of atoms is very small. IR in any substance spends its energy on ionization and excitation of atoms and molecules; about half of the charged particles’ energy causes ionization and the other half excites atoms and molecules of the substances.
In case of low dose exposure, only a small number of atoms are ionized or excited. The excitation energy can also cause biological effects, but they are considered less important than the effects of ionization. Each of the above-mentioned events of interaction involves the transfer of small amount of radiation energy to the exposed substance, which in case of low-LET radiation is usually around 100 eV or less. Such transfers occur in a very short time (less than 10−16 s) but can be widely distributed in the substance in a discrete manner along the tracks. Microdistribution of ionization and excitation events, produced by IR, depends on the type and energy of radiation (Goodhead et al. 1996). For example, the ionization density increases with increasing particle charge and with decreasing speed. It is due to the fact that heavy-charged particles interact more strongly with electrons, and the particles with a lower speed spend more time near electrons and their interaction also turns out to be more powerful. Rarely ionizing radiation, creating several acts of energy transfer per 1 μm track, is known as radiation with low linear energy transfer (LET), such as X-rays or γ-radiation, while radiation, creating a dense ionization along the track, is known as high-LET radiation, for example, α-particles, protons, neutrons, and heavy ions (Kudryashov 2008). Each type of radiation produces certain ionization density and has its own penetrating power which is characterized by different linear energy transfer. For instance, γ-radiation, X-rays, and β-particles are rarely ionizing radiation and have low LET, whereas corpuscular radiation (accelerated heavy-charged particles, neutrons, nuclei of different atoms) is densely ionizing radiation and has high LET (ICRP 2003).
Each track of low-LET radiation, such as X-ray or γ-radiation, which crosses the nucleus of a medium size, results in a relatively small number of ionization events. For example, a track generated by γ-irradiation, which crosses the nucleus 8 μm in diameter, produces an average of about 70 ionization events, which is equivalent to an absorbed dose of about 1 mGy. Individual tracks vary significantly due to stochastic nature and different path length as it passes through the nucleus. Each track of high-LET radiation can lead to thousands of ionization events and result in a relatively high absorbed dose in a cell, for example, 4 MeV α-particle track induces an average of about 23,000 ionization events (370 mGy) in the nucleus of a medium size. However, even under exposure to low LET, sites of relatively dense ionization occur in the nucleus, for example, where low-energy secondary electron comes to a complete stop (Kudryashov 2008).
Energy transferred to a small volume of the substance, such as a cell, is not the same for all atoms and molecules. The average energy deposited per unit mass of the substance is defined as the absorbed dose. Radiation-induced effects in a matter depend on the absorbed dose and microdistribution of energy in the matter, which is determined by the type of radiation.
Processes of absorption, redistribution, and degradation of the energy (ICRP 2005) occur during the initial (physical) stage of ionizing radiation effect on biological systems (the duration of the stage is 10−16–10−13 s). The energy of photons or charged particles absorbed by a living system is fully (directly or indirectly) spent on excitation and ionization of atoms and molecules. At the same time the probability of energy transfer to the molecule depends not on its chemical structure, but on the total electron density. Hits, i.e., interaction events between absorbed energy and the elements of the substance, are not interdependent and follow the Poisson distribution.
Electrons, which are released during ionization under the influence of IR, are of particular interest from a biological point of view, as they cause the polarization of water molecules, and stabilize to a state of hydrated electrons that are able to diffuse over long molecular distances and interact effectively with molecular oxygen and other molecules.
Energy transfer to substance leads to subsequent physicochemical processes such as the free radical formation, which also occurs within a short period of time (10−12 s). Active radicals can move quickly in a substance at some distance from the site of the initial event and cause further chemical changes in the molecules of the matter before they become inactive (within 10−6 s or less). Molecular changes that reflect chemical bond breaking can occur at various time intervals and in different ways, depending on the nature of the medium (ICRP 2003). Thus, radiation tracks can release energy directly into DNA (direct effect), or to molecules which are located in the immediate vicinity of DNA, can be subjected to ionization, and form free radicals that damage DNA (indirect effect).
In a cell, the indirect effect is realized on very short distances of a few nanometers, as the diffusion path of the radicals is limited by their high reactivity. Studies with the administration of radical scavengers into cells show that direct effect accounts for about 30–40 % and indirect for the rest 60–70 % of the radiation effect. It is assumed that initial DNA damages as a result of direct or indirect effects are similar. It is shown that radical ions, induced by direct DNA ionization, may enter into subsequent reactions forming radicals of DNA which are similar to those that appear as a result of the radical water molecule attack.
An important achievement of the radiobiology was the establishment of the role of activated (due to radiolysis) water and various oxidative radicals in the development of biological effects of IR (ICRP 2003, 2005). Toxic agents in the form of ROS are constantly formed in a cell as a result of oxidative metabolism. ROS are generated in the mitochondria and cytoplasm of mammalian cells in vivo. On the average 109 ROS are formed per cell per day. ROS induce various oxidative reactions in a cell; damage DNA, lipids, and proteins; and change intracellular signaling and gene expression. Endogenous ROS mainly cause the development of simple DNA oxidation products (adducts) in a small amount – on the average 10 adducts per second per cell genome. On the average one cell has about 106 oxygen DNA damage per day (ICRP 2003).
It is established that oxygen plays an extremely important role in the radiation cell damage. After the exposure of a cell, it determines the formation of excessive levels of ROS, nitric oxide, peroxynitrites, products of lipid peroxidation (PLPs), toxic oxidative DNA adducts, and others (Kudryashov 2001). Most of the free radicals that interact with DNA in exposed cell emerge as a result of water radiolysis. It is known that water is a significant part of the living organism weight, and both small and large cell molecules are located in the aquatic environment. Moreover, a cell contains two types of water: bound with macromolecules, including DNA (hydrated), and not connected. One DNA nucleotide contains about five bound water molecules. The transfer of IR energy to the water molecules leads to the excitation of the atoms, the ionization of the molecules, and the development of a cascade of reactions with the formation of highly reactive species. These first of all include ionized molecules of water, hydroxyl radical, hydrated electron, hydrogen peroxide, superoxide anion, singlet oxygen, hydrogen, and oxygen radicals, and carriers of unpaired electrons. Although the ionization of water molecules forms active particles, mainly ROS species, arising from the bound water molecules, can interact with DNA. As free radicals in cells diffuse over very small distances (a few nanometers), DNA damage is possible only due to radicals emerging in its immediate vicinity.
All cellular components can be directly attacked by the various water- and fat-soluble pro-oxidants, but the largest effects are due to hydroxyl radical (ICRP 2003, 2005). Hydroxyl radical is a highly active oxidizing agent, which is able to damage virtually any neighboring molecule of the cell. While influencing thiolic proteins, it denatures them and inactivates enzymes. In nucleic acids hydroxyl radical reacts with bases, it damages genome, and also destroys carbohydrate bridges between nucleotides, inducing DNA and RNA strand breaks. Interacting with biomembrane, hydroxyl radical penetrates the lipid layer and initiates chain reaction of lipid peroxidation. However, since hydroxyl radical has a short half-life, it can diffuse only for one or two molecular diameters to interact with cell components. Therefore, the probability of its direct interaction with nuclear DNA is small. Diffusing “hidden” form of hydroxyl radical is hydrogen peroxide (H2O2), which has a longer half-life and can be found in relatively large quantities in a cell. A molecule of hydrogen peroxide “delivers” hydroxyl radical to chromatin and membrane structures. Hydrogen peroxide and ROS are also important mediators of such phenomenon as intercellular apoptosis (Bauer 2007).
Recently, it has been established that a simple chemical compound, nitric oxide (NO), which is enzymatically formed in the body, performs the function of a universal metabolism regulator (ICRP 2005). Apparently, NO is one of the most ancient and universal regulators of intra- and intercellular signaling systems and is also connected with nonenzymatic oxidase processes. Currently, there appear more and more new data on the regulatory function that nitrogen monoxide performs in oxidase processes and in mechanisms of gene expression and protein biosynthesis (Goodhead et al. 1996; Menytsikova et al. 2000; ICRP 2005).
Small size of the diatomic NO molecule and absence of the charge provide its high penetrating power through plasma and intracellular membranes. Nitrogen oxide molecules are relatively long-lived and easily diffuse into biological substance. Nitrogen oxide is highly reactive; it is capable of both activating and inhibiting free radical chain reactions. It has been found that NO synthase is activated in tissues of γ-irradiated animals, which leads to accumulation of excessive nitric oxide. It was shown that in exposed organism this process is initiated by ROS, probably through activation of protein transcription factor NF-kB. Diffusing NO molecules play an important role not only in radiation pathology but also in pathologies of various genesis. Besides, long-term generation of nitric oxide initiates apoptosis and is a key factor of inflammation, carcinogenesis, and development of stress and adaptive reactions (ICRP 2005).
Cytotoxic and cytogenetic effects of nitrogen monoxide appear due to formation of extremely active oxidant – peroxynitrite – produced by the reaction of nitric oxide and superoxide anion radical. It was established that peroxynitrite, diffusing in the cell and penetrating through membranes with anion carriers, causes DNA strand breaks and oxidation of the DNA bases, nitration of guanine and proteins, oxidation of biomembrane lipids, etc.
Formation of reactive free radicals in irradiated cells represents the development of a strong oxidative stress. Presence of the oxygen in the internal medium promotes increase in reactive oxygen species yield and thus enhances radiation effects.
In intact cell oxidative processes are under strict and extremely diverse control of enzymatic and nonenzymatic antioxidant systems; therefore, the natural oxidase reaction rate is slow and natural pro-oxidants (primary products of water radiolysis, ROS, NO system, and PLP) remain at a low stationary level. Antioxidants under normal conditions largely protect macromolecules from the ROS attacks. Antioxidants regulate the level of ROS, RNS, and PLP (ICRP 2000). They also act as endogenous “scavengers” of free radicals or “quenching agent” of excited states of molecules in the irradiated cells, thus reducing the level of radiation damage to DNA and other macromolecules. Antioxidants are also able to reduce the initial level of damage, restoring ion-radical states in local DNA sites, caused by direct ionization. Antioxidants can repair each other and form antioxidant cell systems.
Antioxidants comprise a group of enzymes that form a single metabolic chain that converts oxygen radicals and peroxides to nontoxic metabolites (water, alcohols, etc.). The main antioxidants are enzymes: catalase, superoxide dismutase, glutathione peroxidase, phospholipid glutathione peroxidase, and glutathione reductase (ICRP 2005). The most important intracellular antioxidants, which regulate the metabolism of ROS, are Mn- and Cu–Zn-dependent superoxide dismutases as well as glutathione.
The most active low-molecular antioxidants comprise the so-called antioxidative buffer. Major antioxidant cell substrates are thiol compounds: glutathione and cysteine. The other most active low-molecular antioxidants are biogenic amines (serotonin, histamine, catecholamines, corticosteroids), vitamins (ascorbic acid in the cell cytosol, tocopherol, β-carotin and other carotinoids, localized in biomembrane lipids), and other antioxidants (phospholipids, ubiquinone, urates, bilirubin, phenols, microelements, metal ions of mixed valence).
Introduction of antioxidants into cells before exposure to IR helps to significantly reduce the level of radiation-induced oxidative stress and the amount of DNA damage, as well as eliminate the effects associated with these lesions (ICRP 2005). Therefore, the balance of the content of intracellular oxygen and antioxidants-”free radical scavengers” plays a very important role in the formation of DNA radiation damage. After the exposure, the cells and tissues undergo changes in the ratio of stationary level of antioxidants and pro-oxidants.
3.1.2 Primary Lesions of DNA
Advances in biology and first of all in molecular biology in recent years have contributed to a deeper understanding of not only physicochemical but also biological effects of IR. Currently it was proved that it is the nuclear DNA that is the major cellular target of IR. Radiation-induced DNA damages are of complex and clustered character. Total-body exposure with low-LET radiation at a dose of 1 mGy is capable to produce on the average one track in each cell. A dose of 1 mGy per cell results in a 0.04 simple double-strand breaks (DSB) and about 0.001–0.002 complex double-strand breaks, i.e., one DSB is formed in one out of every 25 irradiated cells and complex DNA damage in one out of 500–1,000 cells.
A considerable part of the radiation-induced DNA damage is a combination of double-strand and single-strand breaks (SSB), base damages, and also the sugar–phosphate residues of DNA, alkali-labile sites, DNA–DNA and DNA–protein cross-links, and others, located densely enough to form clusters. If we consider DSB, SSB, and base damages taken together, then complex clustered damage after exposure to low- and high-LET radiation can reach 60 and 90 %, respectively (ICRP 2008).
Ionization results in damaged chemical bonds in DNA molecules. If most of ionization events are single and not related to each other (in case of exposure to low-LET radiation), then the breaks will be easily repaired by cellular enzymes. However, ionization density of high-LET radiation is such that when a particle crosses DNA duplex, it can induce several ionization events. That is why a large amount of damage from high-LET radiation and small from low-LET radiation occur due to localized ionization clusters, which can significantly impair DNA structure. Tracks from high-LET radiation more efficiently induce larger ionization clusters and therefore cause more complex damage (ICRP 2003, 2005, 2008). Thus, high-LET radiation effect is capable of creating very large ionization clusters, which low-LET radiation does not form. As a result, in case of high-LET radiation, the damage can be irreparable and cause severe consequences for the cell. Besides, if the cell has been exposed to high-LET radiation, each track produces a large number of ionization events, so that the cell receives a relatively high dose. On the contrary, the effect of low-LET radiation is more evenly distributed over the cell population. Thus, at doses of low-LET radiation >1 mGy (mean cell nucleus diameter – 8 μm), every cell nucleus would be crossed by more than one rarely ionizing track.
IR causes a lot of structural damage in the DNA of irradiated organisms. At present time, more than 100 products of DNA radiolysis and structural defects of DNA in irradiated cells are known. Radiation damage can affect any of DNA components. Its formation is influenced by the degree of cell oxygenation, level of endogenous antioxidants, protein complexing of DNA, DNA compaction in chromatin, and the activity of DNA repair systems. From a biological point of view, the most interesting are the structural damage of polynucleotide chains and modification of bases and desoxyribose, which appear in DNA of exposed cells. It was established that DNA damage products themselves can affect various cellular structures (Gaziyev 1999).
Some radiation-induced DNA damage is similar, in terms of chemical structure, to spontaneous damage caused by thermal DNA instability, and endogenous oxidative and enzymatic processes. A number of metabolic reactions in the cell are associated with the formation of oxidative radicals, which can attack DNA, causing base damage and DNA strand breaks, but in most cases these events are rare. Complex clustered damage cannot occur spontaneously, as high local concentrations of endogenous radicals are unlikely to appear in close proximity to the DNA.
The major spontaneous damage is depurination (from 2,000 to 10,000 events per human cell per day) and depyrimidination, as well as deamination of the bases. Apurinization and oxidative modification of bases are most active in vivo. Oxidative modification of bases in the DNA of normal cells results from the effects of various ROS. Spontaneous damage is repaired quite effectively in a cell. DNA of each human cell eliminates up to 20,000 oxidation products a day. If this damage is not effectively repaired, then its accumulation can result in mutagenesis and genetic instability, carcinogenesis, and development of degenerative processes.
Spontaneous level of damaged molecules accounts for a few percent of the total amount of DNA. However, it is unlikely that mammalian cells have a high stationary level of DNA double-strand breaks, as these breaks serve as signal for damage recognition processes, which may block the cell cycle or induce apoptosis. Even a single unrepaired double-strand break is able to trigger the above-mentioned processes. That is why it is possible to assume that cell tolerance to DNA double-strand breaks is quite low.
IR interaction with DNA leads to damage of various types, and the resulting chemical products in many cases are identified and then classified according to their structure (Gaziyev 1999). These products vary in damaged chemical bond, modified base, and the extent of damage within a given DNA segment. Nominally all the radiation-induced DNA damage can be divided into two groups (ICRP 2003, 2005, 2007). The first group includes single (one-site) damage: the modification of bases, single-strand breaks, and “alkali-labile” sites (including those devoided of their bases). The second group consists of multiple local damages that include clusters of single lesions in local DNA site, double-strand breaks, and intermolecular cross-linking. Radiation output of the second group of damage increases with increasing LET of the radiation, and these lesions predominate in radiation death, appearance of chromosome aberrations and gene mutations, and cell transformation.
Not only antioxidants and amount of oxygen but also proteins, strongly bound with DNA, can influence the formation of DNA damage in exposed cells. It is well known that in mammalian cells DNA is complexed with histones and non-histone proteins to form highly ordered compact structure (ICRP 2003). Ionization, which occurs at the initial radiation energy transfer in the atoms of the DNA molecules, can be retrieved through strong DNA binding with histone and non-histone proteins of chromatin. However, DNA packing density in different parts of the chromatin varies. Only a small part of the chromatin is relaxed, it is composed of DNA containing active transcription genes. Polynucleotide chains in relaxed chromatin sites are more susceptible to the effect of IR, a variety of chemical agents and enzymes of DNA metabolism. The other large part of the chromatin is highly condensed heterochromatin which is not able to be transcribed, and its DNA is represented by sequences, which are not encoded, or “silent” genes and is shielded by proteins that protect them from the effects of different damaging agents. Therefore, the heterogeneity of the DNA packing geometry in the chromatin and its complexing with nuclear proteins are important, both for the formation of DNA radiation damage and for the access of DNA repair enzymes to these damaged sites (ICRP 2003). So DNA decompaction in the structure of chromatin and its total deproteinization prior to the exposure have a significant effect on the formation of DNA radiation damage. The number of different DNA lesions, estimated per unit dose at irradiation in solution (in vitro), is about 60–100 times higher than the number of the same damage, registered in the DNA of irradiated cells in vivo. This confirms that the frequency of DNA damage in more open, actively transcribed regions of chromatin is much higher than in the fragments of the same length in the DNA of inactive heterochromatin.
The results of many studies demonstrate that IR induces various chemically stable structural damage in the DNA of irradiated cells. According to the estimations, the radiation output of DNA damage is a linear function of the exposure dose. Much of this DNA damage in irradiated cells undergoes rapid enzymatic repair; that is why the level of the damage registered immediately after irradiation also depends on the dose rate (ICRP 2005). Thus, complete formation of DNA structural radiation damage, crucial for mutagenesis, transformation, and cell death, depends not only on the dose and physical characteristics of radiation exposure but also on the ability of cells to prevent chemical fixation of the initial damage and to recover with the help of enzyme systems.
Typically, the radiation output of stable DNA lesions can be adequately registered immediately after the exposure of the cells in case of DNA repair enzyme inhibition (Gaziyev 1999). The γ-radiation effect causes the formation of about 3–6 chromosome breaks (determined by the method of premature interphase chromatin condensation), 20–60 double-strand breaks, about 200–400 local clustered lesions, 800–1,000 single-strand breaks, 150 DNA–protein cross-links, and 250 DNA base damage (including base modification and base excision from the DNA) (the estimation was performed per 1 diploid genome).
Not only DNA damage but also DNA protection can be induced by endogenous toxins (such as ROS). For example, relatively low and moderate concentration of ROS can cause the formation of DNA oxidative adducts; they can also perform signaling function to induce adaptive responses which can result in the increase of cell viability. High concentrations of ROS lead to the damages that initiate cell necrosis (ICRP 2003). Apparently, cells do not differentiate endogenous ROS and those produced by low doses of IR. The nature and intensity of DNA damage as well as protection depend on the type of radiation, cell type, cell cycle, and metabolism. As it has already been mentioned, physiological defense mechanisms include elimination of toxic agents (especially ROS) by means of various biophysical and biochemical systems and cell removal through apoptosis, necrosis, or immune response. Thus, on the one hand, ROS can cause damage to cellular components, especially DNA; on the other hand, they can act as signals of extremely important cell functions (adaptation, proliferation, and apoptosis). The balance of these effects depends on the concentration of intracellular ROS.
The development of oxidative process in the form of oxidation chain reactions in biomembranes is a mechanism of primary lesions enhancement, which ends in irreversible oxidative degradation of cell membrane structures. Irreversible degradation, both DNA and biomembranes, can have fatal consequences for the cell. Mechanisms of oxidative degradation of biomembranes have been thoroughly studied (Kudryashov 2008). They are known to be most susceptible to oxidative degradation as unsaturated sites of phospholipid fatty acids (such as linolenic, arachidonic) are extremely sensitive to oxidation. Great amount of polyunsaturated fatty acids in the phospholipids determines the high capability of biomembranes to oxidation chain reaction and to formation of PLP that possess oxidase activity. The oxidation reaction of lipids, initiated by IR, plays an important role in pathology and death of the cell. The significance of such reaction is in its contribution to the large-scale accumulation of excess toxic PLPs (hydroperoxides, epoxides, aldehydes, ketones), through which IR also indirectly affects DNA.
3.1.3 Cellular Response to Low Dose Rate Radiation
A cell is the basic structural unit of tissues; it structurally and functionally interacts with neighboring cells and tissue matrix by exchanging signals. The study of cell and tissue reactions to low dose rate has fundamental importance for understanding the mechanisms of adaptation to chronic radiation effects.
It is well known that the most dangerous consequences of cell exposure are caused by radiation damage to their nuclei. With the use of IR microbeams, which are able to irradiate separate cell components, it was proved that it is the nuclear DNA which is the major cellular target for IR (ICRP 2008). Ionization of DNA molecule can result in mutations, in chromosome aberrations (CA), and in the loss of mitotic ability (cell death).
As it has been mentioned above, many DNA lesions result from direct effect of IR, ROS, PLP, and nitric oxide system. Initial DNA damage triggers various defense mechanisms aimed at compensatory mechanisms mobilization and activation of damaged structures reparation. These defense processes are of special importance for maintaining the structure and function of critical structures – targets. After the irradiation, the cell defense mechanisms are triggered by at least two ways. Some mechanisms involve rapid responses, such as cell damage recovery (e.g., DNA), which can take from several minutes to several hours after exposure, depending on the nature of the damage. Others start with a delay of several hours after low dose exposure. It induces reactions similar to stress, which can last up to several weeks, and provide greater protection against ROS, and other active radicals.
The assumption that DNA reaction activity to damage and its repair process are the main determinants of the dose, dose rate, and radiation quality effects at the cell level was confirmed in different studies of the cells (ICRP 2008).
One of the early specific changes in cells, observed after the exposure to IR, is the induction of certain protein kinases. IR induces activation of both p44/42 MAPKs and SAPKs. Kinases transmit signals, induced by nuclear DNA damage, and regulate early response gene expression (jun, fos, egr), which encodes related transcription factors. Transcription factors, produced during the early response to IR, can bind (limit) promoter regions of several genes and consequently increase the synthesis of new copies. Although both enzymes are induced after the irradiation, MAPKs are activated predominantly by some growth factors and phorbol esters, whereas SAPKs by UV or inhibitors of protein synthesis. Radiation-induced SAPKs activation is absent or reduced in AT cells, which are more sensitive to DNA damage. IR also induces p53, PKC, PK50, and PK55 (Stecca and Gerber 1998; Sasaki et al. 2002). More than 150 human genes are known to be involved in DNA damage repair.
Signaling pathways, activated by IR, involved in cellular response to damage, induce cell cycle delay and DNA reparation. The first consequence of transcription activation, induced by p53, is the cell cycle block in checkpoints to prevent the multiplication of the appeared DNA defects by means of DNA reparation (Sasaki et al. 2002).
It is known that in the course of the cycle, a cell undergoes several discrete transitions, normally performed due to the regulatory mechanisms in strict order and at the correct time. Cell transition to a new stage cannot occur if not all the processes, inherent in the preceding stage of the cycle, were completed. Each stage of the cell cycle is under checkpoint control (Elledge 1996; Zhou and Elledge 2000). They regulate the order and time of cells transition from one stage to another and ensure that such critical phenomena as DNA replication and chromosome segregation complete with exact accuracy. Checkpoints respond to the damage by cell cycle block, in order to provide sufficient time for reparation and induction of genes that accelerate reparation. Loss of checkpoints in multicellular organisms results in genome instability and uncontrolled proliferation and promotes malignant transformation of normal cells.
Cell cycle delay in irradiated cells, biochemically bound to complex signaling system at DNA damage, makes it possible to increase the efficiency of reparation up to its maximum or leads to cell death. Cell cycle block after exposure to damaging factors (including IR) is carried out by blocking the signaling pathways of proliferation (Nyberg et al. 2002; Satyanarayana et al. 2008). The system of checkpoints is the basic system that arrests cell cycle in response to radiation-induced damage. The system of checkpoint proteins detects DNA damage and provides adequate cell response to damaging factor (Zhou and Elledge 2000). It was shown that checkpoint proteins control the integrity of DNA structure, the accuracy of DNA replication, completeness of each stage of the cell cycle, and cell preparedness for the next stage (Elledge 1996; Kops et al. 2005). In case of DNA damage, the mechanisms of cell cycle block are triggered in the cell (Edwards et al. 2002). Thus, the cells exposed to γ-radiation are blocked at the stages G1 and G2. It was established that in addition to cell cycle block that can initiate the radiation-induced DNA damage, checkpoint proteins can also activate reparation. If the damage is not repaired, then they can activate apoptosis (Nyberg 2002).
DNA reparation begins almost immediately after the damage occurrence. Some changes of the bases restore within 10 min–1 h. The period of partial recovery of DNA single-strand breaks makes up less than 10 min. and DSB more than half an hour. DNA reparation systems function in the cell is to repair DNA damage. Some of them are activated by DNA damage itself, and their activity depends on the absorbed dose. The process of DNA structure damage recovery with reparation enzymes is preceded by chromatin transformations or translocation of chromosomes loci in the cell nucleus.
As it has been noted, DNA damage reparation is the crucial process in the development of the biological radiation effects after low dose/low dose rate (LD/LDR) exposure. It is shown that incomplete reparation or reparation with errors of DSB best of all explains the cell reactions to irradiation (induction of chromosome aberrations and gene mutations, cell death). DNA double-strand breaks induced by low exposure doses (about 1 mGy) remain unrepaired for many days, as compared to a more efficient DSB reparation, which is observed at high exposure doses.
Initial DNA damage itself (SSB and DSB, damage and/or release of bases) is not dangerous for the cell. However, their inefficient reparation and subsequent DNA replication result in mutations that pose a real danger to the cell viability. If the direct initial radiation damage is not repaired and is localized in the structural part of the gene (mutation), then its expression will lead to synthesis of hundreds and thousands of mutant protein molecules that violate cell functioning or cause cell changes that are incompatible with its viability. The next stage of the initial DNA damage amplification may be associated with their transfer (inheritance) to “mother cell” offsprings, i.e., the number of cells carrying damage increases. Thus, the impairment of DNA chemical structure is the most important primary stage of mutagenesis for the future of the cell. The intensity of mutagenesis depends on the genotype of an organism, which determines the activity of repair and antioxidant systems.
Despite the high efficiency of repair systems, some initial damage avoids recovery and induces persistent DNA damage. Primary DNA damage is fixed in the form of mutations in the process of DNA reparation and replication. Improperly repaired or unrepaired lesions can make viable daughter cells more susceptible to oncogenic transformation or cause genomic instability. Such cells are more sensitive to repeated attacks of active radicals and are prone to elimination by the immune response or apoptosis. Removal of damaged cells by apoptosis occurs within a few hours after exposure.
Apoptotic elimination of cells with radiation damage, which leads to the removal of cells with mutations, is considered to be an alternative to reparation. Violations of coherence in DNA damage reparation processes, on the one hand, and apoptosis and cell cycle control, on the other, can lead to genomic instability and malignant transformation of the irradiated cells.
There is a strong bond between the biophysical processes, which determines the induction of complex initial DNA damage, and cellular responses to DNA damage, on the one hand, and frequency of gene mutations and chromosome aberrations after the exposure, on the other. Moreover the shape of dose–mutation effect function is determined by biological system, mutation endpoint, radiation quality (LET), and dose rate (UNSCEAR 1993, 2000). Dose–effect function has linear–quadratic form for low-LET radiation and is nearly linear with increasing LET. For low-LET radiation decline in dose rate reduces the frequency of gene and chromosome mutations in somatic and germ cells of mammals. Maximum values of the dose rate factor reach 3–4.
Besides, absolutely new phenomena, induced by low doses, were discovered. It was proved that cellular responses to LD/LDRs are fundamentally different from responses to high doses and high dose rates both qualitatively and quantitatively. These are mainly delayed and transient cellular signaling changes that affect intercellular enzyme activity, cell response to ROS, DNA synthesis and recovery, apoptosis, cell differentiation, and immune response. These adaptive responses occur due to changed gene expression through up- or downregulation of the genes, which are responsive only to low doses. Specific cellular response to low doses should be considered in the context of other recently established phenomena that may occur after single cell exposure to high doses. These reactions include the so-called bystander effect, genomic instability, and epigenetic effects that can occur with the cell offsprings over many cell generations (Feinendegen et al. 1996).
It is well known that the behavior of a single cell or tissue is determined by protein expression. Proteins are encoded by genes whose expression is regulated at many levels by epigenetic modifications, such as DNA methylation, and binding of transcription factor and micro-RNA. Proteins perform structural, enzymatic, and signaling functions, and the function of the protein can be regulated by a number of chemical modifications, such as phosphorylation. Cell and tissue exposure can change gene expression and protein expression and also can cause a modification of the protein, gene, and chromatin, which will have an impact on cell function. Gene expression can be modified by epigenetic alterations of DNA and chromatin. It was demonstrated that acute and chronic low-dose exposures in vivo affect DNA methylation depending on the tissue, sex, and dose rate (Kovalchuk et al. 2004). Radiation can also influence histone methylation profiles (Pogribny et al. 2005).
It was proved that low doses of IR can induce increased radioresistance of cells to subsequent exposure to higher doses (Savant et al. 2001; Pelevina et al. 2007). This effect acquired the name of radioadaptive response. Induction of adaptive responses involves several systems, including nucleotide excision repair, nonhomologous end joining (NHEJ) of DNA strands, antioxidant defense mechanisms, and such factors of cell cycle as cyclin D1 (Hafer et al. 2007; Fan et al. 2007; Otsuka et al. 2006; Ahmed et al. 2008). Certain data shows that some of the adaptive responses are induced by the mechanisms of bystander effect (Klammer et al. 2010). It is possible to assume that radiation-induced genomic instability, bystander effect, and adaptive response are interconnected, and it was proved that they can develop in some common ways. It is important to point out that the adaptive response (AR) of the lymphocytes is characterized by considerable individual variability. Moreover, not all the individuals showed AR. The induction of AR is influenced by the age of the individual, environmental conditions, incidence of somatic and genetic diseases, immune status, stress, physiological state of the organism, and genotype (Pelevina et al. 2007).
It was demonstrated that cellular response to radiation exposure can result in genomic rearrangement and/or cellular effects without DNA damage (ICRP 2005). Such processes acquired the name of epigenetic and are subdivided into radiation-induced genomic instability (RIGI) and postradiation effect of signal transmission to neighboring cells (“bystander effect” – BE).
RIGI describes genome damage in offspring cells after a great number of cell cycles and is revealed in the increase of the frequency of chromosome aberrations, mutations, apoptosis, and others (Pelevina et al. 1996; Mazurik and Mikhaylov 2001). It is assumed that RIGI is expressed primarily in the genetically modified cells. After in vivo irradiation of a person and mice, cytogenetic results give no evidence of RIGI incidence in hematopoietic cells.
It is assumed that this process involves the mechanisms of cellular stress and oxidative processes. Recent studies of gene expression changes, associated with radiation-induced transmissible instability, presuppose the involvement of multiple mechanisms. It was shown that reparation of the base excision is one of the most effective means of RIGI prevention, as it indicates that SSB and oxidative damage of the bases are key factors of radiation-induced transmissible instability (Somodi et al. 2005). Thus, reduced ability to repair bases and single-strand DNA damage can promote RIGI. This study rejects the assumption that it is the DSB which is a critical event for the instability transmission, but it also suggests that telomeric function is involved in the process. It is assumed that genomic instability can be caused by low dose radiation with low LET, but there is more evidence that doses <0.2 Gy do not induce RIGI. In addition to direct DNA damage, chromatin-bound epigenetic modification contributes to the activation and maintenance transmissible instability (Barber et al. 2009). Finally, there is evidence that radiation-induced dysregulation of centrosomes can promote genomic instability (Maxwell et al. 2008). Experimental systems make it possible to suggest that radiation can cause transmissible instability in vivo. However, clear evidence has not been obtained.
Bystander effect is registered by the increase in the frequency of chromosome aberrations, gene mutations, cell death, and genomic instability in cells neighboring to irradiated ones. The largest part of BE studies is connected with the exposure to α-particles and protons with high LET, although several studies were conducted with the use of low-LET radiation. Mechanisms of signal transmission to the cell bystander are determined both by direct intercellular communication and by signaling molecules. As in the case of transmissible genomic instability, a lot of mechanisms contribute to bystander effect. But in contrast to transmissible instability, there is certain data on participation of the DNA double-strand breaks reparation, NHEJ pathway in particular, in the development of mutations induced by BE. However the contribution of DNA-PKcs to signaling of DSB, induced by bystander effect, raises some doubts. A number of potential bystander signal mediators was detected, including ROS, nitric oxide, TGFβ, and some other markers of inflammatory response (Burdak-Rothkamm et al. 2007; Shao et al. 2006; Yang et al. 2007; Han et al. 2007). Calcium-mediated signaling can participate in signal transduction from external medium to bystander cells. It was found out that p53 and NF-kB responses contribute to the induction of BE (Ghandhi et al. 2008).
Clear evidence of the existence of signal transmission to cell bystanders on long distances were discovered as a result of in vivo studies of DNA damage and DNA methylation in the skin (Koturbash et al. 2006) and spleen (Ilnytskyy et al. 2009). Some of these phenomena are best defined as abscopal, i.e., effects that are observed outside the irradiated sites of the body. Systemic responses of an organism can also make certain contribution to such effects. Until now no evidence was found that nontarget radiation effects can be associated with radiation pathology.
Nowadays it is clear that IR can affect not only cells but also tissue microenvironment, and this process involves oxygen, inflammatory processes, and TGFβ signaling (Barcellos-Hoff et al. 2005). Such microenvironment modifications can consequently result in changes of the cell growth and normal tissue function.
At certain exposure dose (threshold dose), a significant amount of cells in tissue can die or change their functional activity to the extent that would be sufficient enough for the development of certain tissue or organ failure. That is why dose dependence of radiation-induced tissue reactions in adults and children has true threshold value, which means absence of the risk of their formation at low doses.
3.2 Radioadaptation Under Chronic Exposure to Low-LET IR
Not considering the theory of life and man origin on earth, it should be noted that for a long time mankind has evolved under natural background radiation, which was not constant in space and time. It presupposed that living systems (including humans) developed adaptive reactions to low doses of IR. Current understanding of radiation effects indicates low probability of the fact that doses below 100 mGy per year can lead to severe tissue reactions, even from the most radiosensitive tissues, such as the RBM, gonads, and lens. Defense mechanisms provide the ability of living systems to adapt to IR in the low-dose range.
The mechanisms and strategies of radioadaptation differ significantly depending on the organizational level of the biological system. Thus, cell radioadaptation aims at implementing the strategy of irradiated cell survival. Radioadaptation of the tissue or organ is focused on maintenance of specific functions of the tissue, organ, or system. Radioadaptation of an individual allows maintaining not only the viability and fertility but also homeostasis of the whole body after the exposure to IR. Ultimately, population adaptation to IR ensures species preservation.
Radioadaptation as part of the overall strategy of living system adaptation to the changing environment is the general biological process which is registered both in vitro and in vivo in cells and tissues of various organisms under the effect of rarely ionizing radiation. Data on the ability to adapt to densely ionizing radiation are limited. One of the important criteria of radioadaptation is the ability of cells to increase radioresistance. Although the increase rate of radioresistance in vivo is not great, radioadaptation allows the cell (tissue) to maintain structural and functional stability and proliferative potential if the background radiation deviates from the optimum values. Adaptation is usually manifested at low doses (less than 200 mGy) (Feinendegen et al. 1996; Joiner et al. 1996).
As it has already been mentioned, there are two stages in the development of adaptation: the first stage (of urgent, but incomplete adaptation) and the subsequent stage of long-term adaptation. Urgent stage of adaptation takes place immediately after exposure and can be realized only on the basis of ready-to-use, previously formed physiological mechanisms (e.g., initial level of antioxidants, physiological functional cell reserve). At the core of the long-term adaptation are reparation, regeneration, and compensatory processes, which are realized on the basis of cellular (tissue) responses (Ikushima et al. 1996; Nogami et al. 1993; Seed et al. 2002a, b).
Chronic exposure is characterized by long-term influence of IR when the processes of cell structure, tissue, and organ damage, on the one hand, and adaptation and recovery, on the other, occur simultaneously. Their ratio in the dynamics of chronic exposure predetermines the final result of radiation responses of different tissues (threshold dose, damage severity, latency period duration, etc.).
All tissue reactions and system response of the organism to chronic exposure of IR in the form of CRS are threshold effects. The threshold is first of all determined by the presence of compensatory-adaptive reactions of tissues. Paired organs (e.g., kidneys and lungs), or organs that have functional subunits (e.g., liver), can survive damage of many cells without developing clinical signs of tissue failure due to substantial reserve capacity and compensatory influence of surviving cells. It is also an important reason for the existence of a threshold dose for tissue dysfunction, especially in case of partial organ irradiation, when critical part of the organ can remain unaffected. If the threshold dose is exceeded, then the severity of organ functional disorders increases with increasing dose. In contrast, if organs have sequential structure (e.g., spinal cord), their functional reserve is insignificant or absent, and the tolerance dose is much less dependent on the irradiated volume.
In addition, it is necessary to differentiate the level of doses inducing adaptive tissue reactions and doses that alter the structure and function of tissues and cause cell death. In the analysis of CRS clinical manifestations, it is necessary to take into account the differences between structural and functional tissue tolerance to radiation. Structural tolerance is determined mainly by the death of the tissue-forming cells, whereas functional tolerance by the change of specific activity of the most radiosensitive cells. In this case, the ability of irradiated tissue to maintain its function varies considerably depending not only on the organ dose but also on irradiated volume and structural and functional organization of the tissue.
Tissue reactions, developing as a result of the chronic exposure, have latency period, which is determined not only by radiosensitivity of cells and tissues but also by their adaptive responses. Inadequate processes of adaptation, reparation, and regeneration, preconditioning renewal of the functional cell pool, can lead to structural and functional tissue failure due to the damage caused by IR (e.g., RBM hypoplasia, infertility). The higher the dose and dose rate, the shorter the latency period.
Simultaneous development (combination) of the alteration and adaptation processes under chronic exposure leads to certain periodicity of tissue reactions, such as inhibition of hematopoiesis and immunity (Muksinova and Mushkachyova 1990; Novosyolova and Safonova 1994; Sergeyevich and Karnaukhova 2002) and nervous and endocrine system disorders (Kolomiytseva et al. 2002).
In view of significant dynamics of the alteration and adaptation processes ratio under long-term radiation exposure, it is suggested to differentiate radiosensitivity and radiosensibility of the tissues (Nikolskiy and Koterov 1999). Radiosensitivity and radiosensibility reflect different stages of one and the same unified process of tissue response to chronic exposure. Radiosensitivity can be defined as initial stage of system response (cells, tissues, organs) to irradiation, when adaptive processes compensate for radiation-induced alteration and provide a normal level of its functioning. The main criteria of radiosensitivity can be considered adaptive (compensatory) reactions and degree of their severity, for example, functional changes of specialized blood cells and immunocompetent cells, changes in the duration of cell cycle, and proliferative activity. Radiosensitivity threshold is the minimum dose sufficient for the induction of adaptive reactions. Apparently, doses at which adaptive reactions start to be registered are very low, and they cannot be taken into account for the purposes of radiation safety due to the significant individual variability, but they are extremely important for the analysis of individual radiosensitivity of organism.
Mechanisms of adaptation to chronic exposure in various tissues have certain peculiarities due to structural and functional specificity of their cells (Paranich et al. 2001). Moreover, there is considerable individual variability of tissue reactions to chronic exposure which is preconditioned by certain characteristics of an organism, including radiation damage reparation. That is why under chronic exposure in the doses close to threshold values, tissue reactions as well as stochastic effects are probabilistic in nature. This statement can also be applied to CRS; therefore, while describing threshold dose value, necessary for the CRS formation, it would be more appropriate to speak about threshold dose range.
Radiosensibility is a subsequent stage in the development of cells, tissues, organ systems, and the whole organism response to continuing exposure, which is characterized by changes in homeostasis of cellular and tissue structures. Adaptive (compensatory) processes in this period are not able to provide normal level of tissue, organ, or system functioning. Radiosensibility reflects the severity of radiation tissue damage, which is caused by cell death (due to clonogenic death or apoptosis), functional radiation effects (due to violations of the intra- and intercellular signaling), and also secondary (reactive) responses (Denham et al. 2001).
Evaluation criteria of radiosensibility can be noticeable signs of tissue alterations accompanied by decreased functional activity of the system. Thus, radiosensibility can be defined as a set of decompensated functional and structural disorders in a tissue, organ, or system under the long-term exposure (e.g., cytopenia, RBM hypoplasia, or lymphoid organs). The minimum exposure dose, which leads to the development of both structural and functional changes in tissues, is defined as radiosensibility threshold (Nikolskiy and Koterov 1999).
Currently it is obvious that the adaptive reactions of cells and tissues to radiation exposure is not predetermined solely by exposure dose and dose rate. They can be modified by various biological factors (antioxidants, prostaglandins, heat-shock proteins, growth factors, etc.) and be genetically determined (ICRP 2008). While certain modifiers are most effective when given prior to exposure, increasing or decreasing radiosensitivity of tissues, others appear to have an effect at the time of irradiation, and even after it, providing a therapeutic effect against radiation-induced effects.
Despite a great amount of publications, the mechanisms of adaptation to chronic effect of IR are still insufficiently studied. The mechanism of tissue tolerance to chronic radiation, apparently, is quite complex and is associated with a set of subcellular, cellular, system, organ, and organism adaptive response to chronic exposure. Radioadaptation mechanisms at different levels of biological organization occur simultaneously and determine system’s response to irradiation.
3.2.1 Molecular Mechanisms of Adaptation
AM Kuzin was one of the first scientists who attempted to give a systematic explanation of radioadaptation. He suggested that under the effect of low dose IR, activation of the processes that occur naturally in a living organism (stimulation of cell division, growth, and development) is connected with an excited state of molecules as a result of irradiation. While the damaging effect of IR is connected to the ionization of molecules, which leads to a subsequent change in their structure and violation of their functions, in the development of adaptation, the author attached special significance to the possibility of appearance of collective electronic excitations of protein molecules and nucleic acids under the effect of low dose IR (Kuzin 1995).
In his opinion, the effect of low doses reduces the lag period of cell transition from the resting stage to the division stage; increases the speed of cell division cycle; accelerates the rate of DNA, RNA, and protein synthesis; and raises the number of cell population and intensity of cell metabolism. All these changes are associated with some increase in spontaneous level of free radicals in a cell after exposure to low dose IR, which intensifies the above-mentioned normal physiological processes of the cell. The author assumed that the central mechanism of the increase in proliferative activity of the cell under the effect of low doses is the excitation of the protein molecules of the cell membrane receptors. Growth factors, hormones, electromagnetic radiation, and low dose IR are perceived by membrane receptors. As a result membrane receptors are activated and start to develop primary messengers (cyclic nucleotides, polyamines, etc), which trigger the synthesis of second messengers (phosphorylated proteins, calcium ions, nitric oxide, etc.), directly activating gene, through a number of protein kinases, calmodulin, and others. The author assumed that under the effect of low-dose IR, multiple receptors go into an excited state, which makes them more sensitive to specific effectors, and he suggested three mechanisms of receptor molecules excitation:
Direct excitation of the molecules that form the receptor through direct absorption of IR energy
Indirect excitation of the molecules through active radicals
Activation of the natural effector synthesis
Subsequent studies demonstrated that under the effect of low-dose IR, the transduction systems, indirectly through protein kinase C, activate genes, whose products are involved in DNA reparation (Ikushima et al. 1996), which ultimately increases cell resistance to subsequent test irradiation. It is evidenced by the inhibition of radioadaptive response in case of genetic defects of DNA reparation and use of inhibitors of DNA repair enzymes (3-aminobenzamide, arabinosylcytosine). Stimulation of repair enzymes (DNA ligase, DNA glycosylase, DNA polymerase) under chronic exposure in experimental animals is accompanied by increased radioresistance of animals.
Nowadays it is known that low-dose IR can modify the level of cyclic nucleotides (cAMP and cGMP) and potassium-dependent phosphorylation, which increases the activity of protein protection and reparation factors. Low doses can also stimulate cell proliferation due to the growth factor synthesis, the effect of free radicals, or changes in conformational structure of membranes that alter the receptor sensitivity to mitogenic stimuli. Mechanisms of radioresistance under chronic irradiation are implemented at the cellular level in vivo through the synthesis of stress proteins (heat-shock proteins, metallothioneins) and endogenous antioxidants (glutathione, catalase, and superoxide dismutase) and stimulation of DNA repair synthesis and DNA polymerase activity. The effect of new gene expression in the cells (especially of critical organs) is possible under chronic exposure due to constantly forming free radicals that damage DNA.
The outcome of cell irradiation depends on the dynamics of the balance between the processes of DNA damage and reparation. The positive effect of low radiation exposure doses can be achieved when the radiation-induced protective response exceeds the damaging effect. It was shown that after low-dose exposure the amount of produced antioxidants is greater than that which is required to neutralize the radicals induced by irradiation. This elevated production of antioxidants increases cell protection from free radicals formed during the subsequent exposure (radiation fraction) or radicals of normal oxidative metabolism of the cell (Sagan 1989).
According to Sasaki et al. (2002), the mechanism of reparation activation in adaptive response is mediated by signaling pathways: p38 mitogen-activated protein kinase (p38 MAPK), phospholipase C (PLC), and PKC. The signal of radiation exposure (DNA DSB) is passed through the system of p38 MAPK–phospholipase circuitous signaling pathway; it activates p53 through p38 MAPK, which is balanced by ERK and JNK kinases, and Wip1 phosphatase. Activated p53 triggers reparation by means of NHEJ of DNA strands (illegal recombination) and homologous recombination (but the first one predominates) and inhibits apoptosis (Sasaki et al. 2002).
It is assumed that radioadaptive response in all cells, promoting cell survival, is mediated by nuclear factor kB (NF-kB). Currently it has been shown that NF-kB plays a key role in radioadaptive resistance of tumors in case of fractionated exposure. Increased basal activity of NF-kB in some tumors is associated with resistance to radiation and chemotherapy (Ahmed and Li 2008). It is well known that NF-kB can activate a large number of genes involved in stress response, inflammation, and apoptosis. Blocking NF-kB activity can increase apoptosis and decrease the survival of some human cancer cell lineages.
IR induces critical genes (IL-1β, IL-6, TNF-α, CuSOD) through NF-kB. Antiapoptotic function of NF-kB is bound to TNF-α receptor. Cytokine TNF-α, produced by activated T lymphocytes and macrophages, is able to activate NF-kB. Thus, radiation activation of cytokines can trigger subjacent molecules with NF-kB and JNK activation. ATM mediates NF-kB activation. Two stress responding proteins (metallothionein and Ku autoantigen), activated in radioresistant cells, are also regulated by NF-kB. NF-kB-mediated induction of mitochondrial CuSOD contributes to elimination of radiation-induced ROS and triggers signaling system, which leads to adaptive radioresistance. Moreover, NF-kB is able to inhibit JNK-mediated apoptosis (Ahmed and Li 2008).
It was also established that cellular stress activates transcription factor HSF1 and results in transcription of genes encoding heat-shock proteins (HSPs). As a result, cells with increased HSP content become more resistant to various stresses. It is believed that protective function of HSP is determined by their ability to catalyze renaturation and degradation of labile cellular proteins that undergo denaturation and aggregation under stress effects.
Normal mouse fibroblasts were compared to fibroblasts lacking HSF1 gene, synthesizing transcription factor that initiates expression of HSP in response to stress. Some of the cells were infected with special vectors for the expression of constantly active HSF1 or certain HSPs (HSP70, HSP56, HSP27). It was revealed that heat stress (43 °C, 30 min) raised the level of HSPs in normal fibroblasts and increased their survival after γ-irradiation (4 and 6 Gy). In cells lacking the gene HSF1, heat stress did not cause either HSP induction or enhancement of radioresistance, but these effects are manifested after expression of active HSF1 in these cells. Hyperexpression of HSP70 and HSP26 equally increased radioresistance of fibroblasts. The obtained results show that the accumulation of HSP70 and HSP27 in target cells prior to exposure can make them more resistant to irradiation. It is these proteins that are main radioprotectors among the products of HSF1-mediated stress response. Pre-radiation stress response increases cellular radioresistance. It is assumed that HSP70 either directly or indirectly participates in protection of DNA radiation damage reparation. The excess of HSP27 in a cell may reduce the nuclear DNA damage caused by oxidative stress (Malyutina and Kabakov 2007).
It was established that radioresistance induced by HSP70 is connected with increased activity of protein kinase C (PKC). There is some evidence of PKC involvement in radioresistance. It can be assumed that the increased activity of PKC is induced by HSP70 and causes radioresistance. PKC gene expression increases soon after irradiation, and PKC engages in DNA reparation. PKC activates the products of proto-oncogenes Raf and Mos. The expression of these genes is associated with radioresistance. Similarly, activated Ras causes radioresistance and is also associated with increased expression of PKC. It was shown that PKC affects the radioresistance of cells by blocking radiation-induced cell death through apoptosis (Park et al. 2000).
The involvement of p53 in radioadaptation testifies to the fact that living organisms develop an effective system to detect significant genetic stress and for cell response: damage reparation for cell survival or apoptosis in order to achieve a higher level of tissue reparation and avoidance of harmful effects (transformation). In all these processes, p53 is the key factor of signaling system response to stress. It is likely that such life-or-death decision is programmed at the tissue level (Sasaki 2002).
Wolff et al. (1996) showed that DNA reparation can increase after the exposure to low doses. It implies another defense mechanism, namely, irradiated DNA becomes better fit for damage reparation with subsequent exposures (Wolff 1996).
It is possible that radiation factor at low doses, generally producing derepressive effect on the genome, can increase the accessibility of DNA damaged sites for repair enzymes. The presence of multiple intracellular radiation-induced syndromes suggests their possible role in radioadaptation mechanisms. Until present, discussing the multilayered DNA repair system from radiation damage at the subcellular level, we paid special attention to the role of stimulation of the primary DNA structure reparation processes. But the fact of accelerated differentiation of irradiated cells can testify to the existence of epigenetic radioadaptation mechanism. Ultimately, the ability of cells to respond to critical effects and, therefore, under certain conditions, to increase their stability (i.e., to adapt) is determined precisely by the modification of the epigenetic level of the gene activity regulation (Feinendegen et al. 2004).
3.2.2 Mechanisms of Cell and Tissue Radioadaptation
Radioadaptation in a broad sense is aimed at reducing the number of damaged cells and increasing their survival rate, compensation of dead cells, and maintenance of functional status, first of all of the major life support of the body. It is important to mention that the mechanisms of cell and tissue adaptation to chronic exposure have been extensively studied only in recent years.
As it has already been noted, adaptive protection can be observed in mammalian cells both in vivo and in vitro after chronic exposure to low-LET radiation. Adaptive processes at cellular and subcellular levels develop with some delay (hours) after exposure and include detoxification of ROS and other reactive radicals, induction of DNA damage reparation, increased proliferative capacity of stem cells, shortening of the cell cycle period, increased radioresistance of cell populations, removal of damaged cells by apoptosis, and immune responses.
Low doses due to ionization of atoms and molecules can induce the formation of ROS and change the transmission of intra- and intercellular signals that induce adaptive protection mechanisms which except for apoptosis are not observed at high doses. Resulting biochemical reactions develop relatively slowly (within a few hours), may last for several weeks, and resemble physiological stress responses, which protect against DNA damage from any source whether it originates endogenously or from renewed irradiation. Such protective responses appear to depend on mammalian species, types of tissue, cell types, and the cell cycle.
The key mechanisms of cell adaptation to chronic radiation exposure are presented below:
1.
Activation of radioprotective systems (synthesis of endogenous stress proteins, HSP70, HSP72; metallothioneins; antioxidants, glutathione, superoxide dismutase, catalase, and others), which leads to increased radioresistance of cells (including stem cells) (Nogami et al. 1993; Kojima et al. 2002). It is assumed that the induction of the new protein synthesis in the cells of critical organs is the effect of gene expression and is mediated through protein kinase C and nuclear factor κB (Ahmed and Li 2008). Glutathione, produced by the cells after low-dose exposure, also has a stimulating effect on the immune responses (Kojima et al. 2002).
3.
Damaged cells can be removed by changing the intracellular and intercellular signaling (e.g., the one that induces apoptosis). Apoptosis is one of the main pathways of elimination of damaged and abnormal cells from the body. Low doses can induce apoptosis in the cells prior to DNA damage. Thus, chronic exposure, even at low doses, is the basis for the selection of the most radioresistant cells by means of apoptosis; the proliferation of these cells may increase with enhanced radioresistance of the tissue in general (Nikolskiy and Koterov 1999). Cells with unrepaired damage can also be removed as a result of premature differentiation and senescence.
The enumerated mechanisms of adaptive protection involve changes in gene expression. Thus, exposure of human skin fibroblasts in culture to a single dose of 20 mGy γ-radiation caused more than 100 genes to change their expression (including stress response genes) within 2 h. In most mammalian cells examined so far, the expression of adaptive protection from low-LET radiation had a maximum above 5 mGy and below about 200 mGy, and except for apoptosis it significantly decreases at doses >200 mGy (Feinendegen et al. 1996).
It was shown that the selection of more radioresistant cells (Kojima et al. 2002), switching off the apoptosis, and increase of the proliferative activity of cells (including the pool of stem cells and progenitor cells) which can replace damaged or dead cells are of great importance for the maintenance of cellular homeostasis under chronic exposure. In this case, the induction of adaptive responses in cells, excluding apoptosis, decreases with increasing dose of low-LET IR. The various mechanisms of protection may be linked to transient changes in the activity of the G1 cell cycle checkpoint. Detailed description of adaptation mechanisms to exposure in vivo is provided in the study of Nikolskiy and Koterov (1999).
Thus, it seems that adaptive protection is a physiological expression of cellular capabilities to maintain integrity of tissue structure and function in the face of various exposures to potentially toxic agents including IR. In fact, adaptive protection following low doses of low-LET radiation appears to be the consequence of changed cellular signaling and to be universal.
Tissue reaction to irradiation is an integral result of all (various) cellular responses in the irradiated tissue and system, to which this tissue belongs. The function of the entire system (organ) after exposure is determined by the harmonized contribution of the cells constituting the tissue of the given system (organ). Thus, the tissue should be viewed as a complex adaptive system and tissue response to low doses of IR as a result of a variety of cellular responses, including those that are not associated with cell death. Thus, low doses of radiation with low LET can induce apoptosis and therefore eliminate the damage in irradiated cells (Feinendegen et al. 1996).
As it has already been mentioned, the irradiated cells within the tissue can communicate with non-irradiated cells, causing DNA damage in non-irradiated cells via BE. At low doses this damaging BE will cause greater harm than expected under the assumption that damage comes only from directly irradiated cells, i.e., bystander-induced damage amplification appears. In fact, there is evidence that BE also induces protective responses in non-irradiated neighboring cells (adaptive response) and that normal cells in culture may signal for the apoptosis of transformed neighboring cells.
Moreover, the physiological intercellular signaling, also involving tissue matrix function, appears to be less disturbed by low doses than high doses. Cell responses in multicellular systems (and not single cells) include not only intercellular BE of both (damaging and protective) types but also responses to any extracellular signaling. Thus, the tissue as a whole should be considered as a system being composed of elements with different radiation sensitivities and responses to IR but reacting as a whole. Besides it is necessary to take into account microdistribution of dose in tissues, which under low-dose irradiation does not fully reflect average doses to the cells.
The ratio of various nontarget low-dose effects in the tissue has been insufficiently studied so far. However, it is shown that the AR and BE can occur in the same experimental systems. When the cell culture C3H10T1/2 was irradiated by a microbeam with a known number of α-particles, fewer cells survived (65 %) than expected following the linear no-threshold model (90 %) due to BE. However, when the culture was preexposed to a dose of 20 mGy γ-radiation for 6 h prior to α-radiation, the survival rate increased up to 75 % due to the adaptive response (Bonner 2003). Thus, it is clear that AR and BE can have the opposite effect on the dose–effect function. BE enhances the low-dose effect through the damage transfer from irradiated to neighboring non-irradiated cells, while AR induces radioresistance of neighboring cells to subsequent irradiation after the primary low dose. Moreover, the AR can develop in non-irradiated neighboring cells through transmission of factors present in the cell medium exposed to low-dose γ-radiation, although irradiated cells themselves did not develop AR. Adaptive BE is associated with lower basal level of p53, increased intracellular levels of ROS, and increased AP-endonuclease (Iyer et al. 2002).
Adaptation mechanisms implemented in the various tissues under chronic exposure have some peculiar features due to structural and functional specificity of their cells (Paranich et al. 2001). Adaptive reactions are expressed differently in various cells and tissues. Moreover, there is considerable individual variability of tissue response to chronic irradiation, which is due to characteristics of the organism, including peculiarities of DNA radiation damage reparation. The tissue or system capability to maintain the integrity of its function varies widely depending on the irradiated volume and the structural and functional organization of tissue (Michalowski 1981).
Tissues vary significantly in terms of their physiological cell regeneration rate, as well as the dynamics of their maturation, senescence, and death. All these processes determine the radiation exposure response rate of different tissues. It is known that rapidly proliferating tissues have stem cells, which support the departments of proliferating cells as well as of differentiating and mature cells. The timing of the radiation damage appearance in such tissues depends on the life span of mature cells, which are relatively radioresistant. After chronic or fractionated irradiation, increased proliferation of stem cells can compensate for cell death and reduce tissue radiation damage. It should be noted that most of the stem cells normally divide relatively slowly, more slowly than their daughter progenitor cells. The most common rapidly proliferating tissues are epithelial tissues of the intestine and epidermis, as well as RBM cells (ICRP 1987).
In some tissues the existence of stem cells has not been established yet. Such tissues (e.g., liver, blood vessels) demonstrate low proliferative potential and are significantly less prone to compensatory proliferation under chronic exposure (Michalowski 1981). As tissues and organs are made up of a variety of cells with different proliferation rate, the severity of radiation effect within one and the same period of time varies greatly in diverse structural elements of the tissues within the body.
Paired organs (lungs, kidneys, salivary glands) and the organs composed of functional subunits (liver) are characterized by low tolerance to total-body irradiation. However, their partial exposure at high doses does not result in tissue dysfunction. The reason is that these organs have a large functional reserve. These tissues have the so-called threshold volume. If the volume of exposed tissue is less than the threshold, even at large doses, radiation damage does not lead to functional failure of the whole tissue (organ), and only when tissue damage exceeds the threshold, the clinical effects in organ or tissue are induced.
In contrast, in organs such as the small intestine, which have sequential organization, the inactivation of one critical subunit may cause the entire organ failure (Withers et al. 1988). The probability of critical subunit inactivation due to exposure at one and the same dose increases with the irradiated tissue volume. For these tissues the risk of complications is primarily due to high-dose exposure of single sites.
3.2.3 Radioadaptation Mechanisms at the Organismic Level
The responses of the organism as a whole to chronic exposure correspond to classical concepts of general adaptation syndrome (GAS). In response to chronic exposure, pituitary–adrenocortical axis system undergoes changes typical for GAS, which leads to an increase in the synthesis and secretion of adrenocorticotropic hormone (ACTH) and steroid hormones. CNS plays an important role in stress development under chronic exposure (Baraboy and Oleynik 1999).
Long-term (months–years) radiation exposure, even at low-dose rate, may reduce the compensatory-adaptive mechanisms and adaptation failure. Under chronic exposure when alteration processes prevail, the adaptive capability wears out, and radiation damage to cells and tissues accumulates. For example, residents of the Techa riverside villages, chronically exposed to a wide range of RBM doses (0.02–2.05 Gy), in 48–52 years after the onset of irradiation showed decreased ability of blood lymphocytes to the induction of adaptive response (Akleyev et al. 2004). Perhaps this effect is the consequence of reduced efficiency of reparative DNA synthesis under long-term exposure (Nikanorova et al. 2002) or depletion of the cellular antioxidant potential (Ossipov et al. 2002).
In case of normal functioning of the body, the physiological loss of mature cells occurs, but under chronic exposure it may not be adequately compensated for by the cell production due to the decline in the potential of highly radiosensitive stem and progenitor cells. Thus, in the long-term period of chronic exposure, it is possible that the function of the system or organ (tissue) will be impaired, due not only to organic changes (such as BM hypoplasia, vascular changes) but also to the limited possibilities of the physiological tissue regeneration resulting from depletion of the stem cell pool. During the adaptation to chronic radiation exposure, energy, metabolic, and structural resources of the body are redistributed for the benefit of organs and systems that perform essential functions. For example, in the RBM such priority belongs to erythroid lineage that provides for tissue gas transport.
Adaptation of the organism under chronic irradiation is also aimed at the elimination of the surviving but transformed cells, which can lead to the development of malignant neoplasms. Removal of damaged cells occurs in vivo by stimulation of the immune response with low-dose IR. It was established that low doses of low-LET IR can stimulate the immune system, resulting in reduced survival of malignant cells in tissues (UNSCEAR 2009). The selection of more radioresistant cells is important for the maintenance of cellular homeostasis under chronic exposure, while modified radiosensitive cell populations die via immune response and apoptosis. Endogenous glutathione, produced in cells under the effect of low exposure dose, stimulates immune responses (e.g., it increases IL2 synthesis in lymphocytes, which stimulates proliferation and activity of NK cells (Kojima 2002)).
The adaptation strategy of a large group of individuals or population to IR is first of all aimed at the elimination of single individuals, who could potentially develop transgeneration radiation effects, before they reach the reproductive age.
3.3 Tissue Reactions to Chronic Radiation Exposure and Their Role in CRS Pathogenesis
Previously it has been affirmed that reactions of tissues to chronic total-body exposure, the sum of which forms CRS clinical picture, are determined only by the exposure dose and dose rate. Today it is clear that they depend not only on their dosimetric parameters but also on the age, initial tissue status, general organs and body health status, and its genetic properties. Moreover, they can be modified by various biological agents (antioxidants, prostaglandins, heat-shock proteins, cytokines, growth factors, etc.).
As it has already been noted, the peculiar feature of chronic radiation exposure is a long-term development of the effect of cell and tissue damage, on the one side, and compensatory-reparative processes, on the other hand, which occur simultaneously. Their ratio in the dynamics of chronic exposure, ultimately, determines cumulative tissue response to radiation exposure. The concurrent development of these processes leads to periodicity (nonmonotonicity) in the processes of inhibition and functional recovery of certain tissues, organs, and systems under chronic exposure (Muksinova and Mushkachyova 1990; Novosyolova and Safonova 1994; Sergeyevich and Karnaukhova 2002; Kolomiytseva et al. 2002).
It is well known that tissue reactions may be observed in the early and late periods after irradiation, depending on the structural and functional characteristics of tissues. Radiation reactions of a tissue are determined by its cellular composition, cell proliferation rate, and cellular responses to radiation, which may be highly tissue specific. Reduction in parenchymal cell population turnover, modified by the influence of stroma, plays an important role in the pathogenesis of early tissue reactions. Early tissue reactions develop within a few hours–weeks after irradiation and late tissue reactions– months or even years after exposure. The latency period of the tissue reactions is determined by radiosensitivity and adaptive capacity of the tissue.
As it has been mentioned above, under irradiation at doses exceeding the threshold value, CRS did not develop in all exposed individuals. This may be due to different individual radiosensitivity, which is, among other things, genetically determined. However, the proportion of people with extremely high radiosensitivity due to genetic properties is apparently quite small. It is known that among the entire population, only very few people (much less than 1 %) are homozygotes in terms of mutations in repair genes and, therefore, are two to three times more sensitive than the population on the average. Patients with HIV and autoimmune diseases also have increased radiosensitivity (UNSCEAR 2009).
3.3.1 General Regularities of Response to Radiation Exposure Manifested by Cells and Tissues
It is important to note that since 1990, when ICRP Publication 60 was issued, the assessment of threshold dose values for radiation-induced tissue reactions remained practically unchanged. It is still considered that doses of chronic or fractionated exposure have less damaging effects than the same doses of acute exposure. It was noted that the threshold value of the RBM annual dose sufficient for the inhibition of the immunity in case of chronic exposure is 0.3–0.5 Gy. Recent studies have shown that the lens can be even more radiosensitive than it was previously considered. The excess of both cortical and posterior subcapsular cataracts at doses lower than previously expected was noted in several studies. ICRP also pointed out that in case of long-term exposure an individual may develop symptoms of CRS, which manifests itself in the presence of impaired hematopoietic, immune, nervous, endocrine, digestive, and cardiovascular systems (ICRP 2008).
Tissue reactions (TR) are threshold effects. In case when exposure lasts for many years, it is unlikely that severe effects will appear in most tissues at doses less than 0.1 Gy per year. The RBM, gonads, and lens are the most radiosensitive organs (ICRP 1987). ICRP uses the term “threshold dose” to refer to the minimum radiation dose that is required to cause a specific tissue effect, and it is equal to the level of irradiation needed to induce observable effect in 1 % of the exposed individuals (ICRP 2012). At the tissue level, there is a variety of protective mechanisms that determine its tolerance to chronic irradiation and threshold dose for dysfunction. These mechanisms include reparation of sublethal damage, repopulation of tissue cells due to surviving proliferating cells, presence of the functional cell reserve in the tissue, and others that also determine the variability of the threshold value, depending on the type of the tissue.
In most tissues the greater the irradiated volume, the more severe the radiation reactions. Decline in regeneration of the early radiation skin damage together with the increase of the irradiated volume of the tissue is predetermined by the insufficient cell migration from the neighboring areas. In case of late reactions, the effect of irradiated volume of the tissue is connected with the architectonics of the organ.
Reduction of the cell population after irradiation is important for the development of early reactions in epithelial tissues. In some types of tissues, fast cell death after irradiation occurs due to apoptosis, for example, of lymphocytes and acinar cells of the salivary glands. At that, the cell death is understood as the loss of reproductive capacity, but not necessarily the loss of physical vitality or its functions. In other tissues cell deficiency is caused mainly by failure of regenerative stem cells or proliferating (differentiating) cells, which could escape apoptosis. Most types of nonproliferating mature cells do not die during radiation exposure, but as a result of natural aging. Premature aging after exposure may promote some late effects of irradiation. It is important to note that as regards hematopoiesis there is a clear dependence of the level of early tissue damage on the target cell survival. For late slowly developing tissue reactions, the relationship between target cell survival and damage is less clear.
It is well known that tissues vary widely in their rates of physiological cell regeneration, as well as in the dynamics of their maturation, aging, and death. All these cell processes determine the rate at which different tissues respond to radiation exposure. It is known that rapidly proliferating tissues have stem cells, which support proliferating cell compartment as well as those of maturing and mature cells. The timing of the radiation-induced injury in these tissues depends on the life span of mature cells, which are relatively radioresistant. After chronic or fractionated irradiation, proliferation of stem cells can compensate for cell death and reduce radiation tissue damage. Examples of most common rapidly proliferating tissues are RBM cells, the epithelium of the intestinal mucosa, and the epidermis.
Rapidly proliferating hematopoietic tissue has well-defined stem cell compartment (capable of indefinite cell renewal), which gives rise to proliferating cell compartment and compartments of differentiating and functioning postmitotic cells. During chronic irradiation stem cell proliferation can compensate for cell killing and reduce the radiation damage. Bone and cartilaginous tissues, which have no stem cells, are characterized by low proliferative activity of cells, and the timing of their response to IR is dose dependent. Skeletal system response appears after quite a long period of time since the exposure.
Since tissues and organs consist of a variety of cells with different proliferation rates, the expression of radiation damage does not occur simultaneously in all the cell population compartments within a certain tissue. In case of chronic exposure, the manifestation of radiation damage also tends to be modified not only by compensatory proliferation but also by other homeostatic processes (sublethal damage reparation, replacement of cells with lethal damage by repopulation and by migration of intact cells from unexposed areas).
In tissues with low proliferative potential (e.g., liver, blood vessels), there is much less chance of compensatory proliferation in case of chronic and fractionated exposure (Michalowski 1981). Since tissues and organs are made up of a variety of cells with different proliferation rate, the severity of radiation effect is not the same at a definite moment in all structural elements of the tissue within the organ.
Recently, it has been increasingly recognized that the structure of organs and tissues plays a major role in their response to irradiation. Paired organs (e.g., lung, kidney, salivary glands), as well as organs composed of functional subunits (FSUs) (e.g., liver), have low tolerance to total-body irradiation. Their partial (local) exposure even at fairly high doses does not lead to dysfunction. The reason is the presence of functional reserve. These tissues have threshold dose effect. Tissue exposure at high doses that are lower than the threshold value does not cause functional failure of the whole tissue (organ), and only when exposure dose exceeds the threshold, it causes clinical effect in organ or tissue (ICRP 2012).
In contrast, organs with sequential organization (e.g., small intestine) the inactivation of one critical subunit may cause loss of function in the whole organ (Withers et al. 1988). The probability of inactivation of critical subunit with the same dose of irradiation increases with increasing length of the irradiated intestine. For these tissues, the risk of complication is strongly influenced by high-dose exposure of single spots.
Earlier explanation of tissue response to radiation exposure was based on the target model, according to which the severity of injury and the time between irradiation and manifestation of damage depends on the death of the target cells, on their characteristics (radiation sensitivity, repair capacity, proliferative activity, etc.), and on the tissue structure. In some tissues (hematopoiesis, intestines), early tissue reactions, to a large extent, are caused by the death of the stem and progenitor cells, resulting in temporary or permanent deficit of mature cells, which depends on the dose and dose rate. In other tissues (thymus, salivary glands), rapid loss of mature cells occurs via apoptosis. Cell depletion plays a major role in the early desquamation reactions of epithelial tissues after exposure and also in the development of agranulocytosis in case of hematopoietic form of ARS.
Most nondividing mature cells are not killed by irradiation, but their number naturally decreases. Complete denudation of tissues at high doses occurs by the time which is equal to the life span of mature cells and the time needed for production of radioresistant progenitor cells. Stromal tissue produces many growth factors that induce repopulation and differentiation required to normalize the cellular composition of tissues. Recovery terms can be shortened, and the recovery may be more complete if exogenous growth factors were used which further stimulate repair processes in the stroma.
Today, however, it is clear that the effect of cell death cannot explain all tissue reactions (especially functional and late). The presence of nonlethal effects after exposure of cells and tissues is also evident. Cell death cannot explain nonlethal effects after low-dose exposure of cells and tissues. As it has already been noted above, the biological effects of low-dose IR is a set of many interrelated cellular responses to stress induced by nuclear DNA damage (antioxidant synthesis, cell cycle block, reparation, cell proliferation, apoptosis, etc.). In addition to cellular DNA damage, ROS and RNS generated within the irradiated tissue also change proteins, lipids, carbohydrates, and other complex molecules and activate signaling pathways (Denham et al. 2001). Fibrosis may form as a result of the cell death. After exposure fibrosis can develop as a primary reaction. It is quite common late effect, caused by premature aging or accelerated postmitotic differentiation, resulting in excessive collagen production by irradiated mesenchymal cells (fibroblasts, myofibroblasts, smooth muscle cells), but not cell death.
Late consequences of CRS are also based not so much as on the death of the target cells (hematopoietic and nervous tissue) as on the tissue reactions that include the secretion of cytokines and other mediators by damaged cells, impairment of vascular function, development of fibrosis, and dystrophy (Denham et al. 2001). For example, a cascade of cytokines can be induced by radiation and can be stored for a long time prior to the death of a large number of cells and the development of clinical signs of tissue damage.
IR is probably capable of causing not only genetic (gene mutations and chromosome aberrations) but also epigenetic changes associated with alterations in the function of the genes. It should be taken into account that any tissue of a multicellular organism is epigenetically homogeneous cell population, and an organ includes a system of tissues, with each cell being characterized by its own epigenotype. Selection of a particular epigenetic status is predetermined by the specifics of cell genome interaction and internal and external regulating (modifying) influences. Inheritable epigenetic changes differ from mutation changes in that they follow a certain regularity, which is preconditioned by genetic program and by involvement into the process of a large number of cells, which undergo changes in the activity of genes, not in structure. According to current understanding, epigenetic changes are implemented through DNA methylation, histone modifications by acetylation, and regulation of gene expression by means of microRNAs.
As it has been noted above, at the present time there is evidence that low doses of IR may also cause nontarget effects that include RIGI and BE. There is some evidence that the progeny of irradiated cells, which was not exposed to IR, may have signs of genomic instability. It was shown that RIGI can persist for a long time after the exposure (Suzuki et al. 2011).
Thus, inheritable epigenetic changes, apparently, are of a large-scale character and are manifested in persistent changes of the gene expression model. IR can modify the level of spontaneous genetic instability inherited in a cell lineage following epigenetic mechanisms. Epigenetic changes induced by IR are an important link in the overall chain of cellular responses that lead to the formation of a variety of biological effects at different structural levels. The evidence was received that cellular response to low-dose irradiation by the type of adaptive response, cell cycle delay, apoptosis, and other radiobiological reactions may depend on epigenetic variability.
Radiobiological parameters and mechanisms of epigenetic reactions of cells and tissues to low-dose irradiation are presented in a number of reviews (Kadhim et al. 2004; Prise 2006; Little 2007).
Late tissue reactions have not only large latency period which depends on the exposure dose but also a long-lasting formation period. They are divided into general (generic), which are observed as a result of direct damage to the irradiated tissues, and indirect (consequentional), which develop as a result of the early reactions. Late tissue reactions are predetermined partly by low renewal rate and the loss of those cellular compartments, which cells are functional and are able to divide.
Thus, the development of tissue reactions may be due to the activity of cytokines and other mediators from damaged cells, leading to both the disturbance of cell function and cell death. These cellular responses (e.g., cytokine cascades) can be initiated prior to cell death and persist for a long time. Besides, late reactions are caused by the dysfunction of a complex system of intercellular signaling that regulates tissue and organ function (ICRP 2012). Late tissue reactions to a large extent are also mediated by vascular changes and inflammation in the tissues. They are exposure dose dependent and tend to progress.
Related posts:
 Definition, Classification, and Clinical Presentation of Chronic Radiation Syndrome (CRS) Associated with Total Exposure to External Radiation
Pathoanatomy of Chronic Radiation Syndrome During the Period of Its Development and at Recovery Stage
General Conclusions
Definition, Classification, and Clinical Presentation of Chronic Radiation Syndrome (CRS) Associated with Total Exposure to External Radiation
Pathoanatomy of Chronic Radiation Syndrome During the Period of Its Development and at Recovery Stage
General Conclusions
 Diagnosis of Chronic Radiation Syndrome
Diagnosis of Chronic Radiation Syndrome
 Epidemiology of Chronic Radiation Syndrome
Epidemiology of Chronic Radiation Syndrome
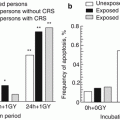 Dynamics of Hematopoietic Changes in Persons with CRS
Dynamics of Hematopoietic Changes in Persons with CRS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


